Chapitre 1:histoire de la chimie
Il est souvent utile d’étudier l’histoire et l’évolution de la science et de voir surtout les étapes les plus importantes de celle-ci dans chaque spécialité. C’ est ainsi que nous allons commencer nos cours sur la chimie. D’après ce qu’on sait, la chimie a commencé avec la découverte du feu qui est essentiellement la combustion d’un réactif pour obtenir la chaleur. Plus tard différents métaux ont été découverts, donnant les noms de fer, de cuivre et de bronze à l’époque de leur découverte.
Cependant nous ne pouvons pas encore parler d’une méthode scientifique à ces moments, c’est plus en relation avec l’évolution. L’évolution est un processus visant à nous adapter au mieux à notre environnement. Ces découvertes ont été plutôt le résultat d’essais et d’erreurs. Les processus conduisant à une meilleure adaptation ont été sauvegardé tandis que les autres ont été abandonnés. Par exemple la découverte du feu a conduit à une amélioration de la vie des hommes pour des raisons évidentes, tandis que le processus n’a pas été très bien compris.
La rationalisation des procédures est d’abord observée chez les Egyptiens (fabrication de verre, de la bière et la coloration),puis les chinois (porcelaine),et les Grecs. Leucippe et Démocrite ont suggéré que la matière était composée de petites particules incassables, les atomos. Les Grecs ont également affirmé que le monde est composé de quatre éléments principaux : la terre, l’eau, l’air et le feu. Nous pouvons maintenant les comparer aux trois phases principales : solide, liquide, gaz et l’énergie
La méthode scientifique a été développée au cours du XVIe siècle. Le procédé consiste en trois étapes :
1) Observation d’un phénomène : donne des informations quantitatives et qualitatives
2) Hypothèse : tente de donner des explications possibles aux phénomènes observés
3) Expériences : rassembler de nouvelles informations sur le phénomène, de confirmer ou non les théories développées dans l’étape précédente.
Avant cela, les hommes ont décrit ce qu’ils ont vu. De ce point, les hommes essaient d’expliquer ce qu’ils voient à travers les théories.
Stoechiométrie et la détermination des masses atomiques :
L’un des pères de la chimie réelle est Lavoisier. Il est connu pour « Rien ne se perd, rien ne se crée, tout se transforme » ce qui signifie que la masse totale des produits d’une réaction est égale à la masse totale des réactifs. Cette déclaration est vrai sauf pour les réactions nucléaires qui convertissent une partie de la masse en énergie.
Joseph Proust a déclaré qu’un composé chimique contient toujours exactement la même proportion d’éléments en masse. Par exemple, dans l’eau pure, la masse de l’hydrogène est toujours 1/9 de la masse de l’échantillon tandis que l’oxygène constitue le 8/9 de la masse.
Pour compléter cette loi, Dalton a observé que lors d’une réaction les masses des composés qui réagissent ensemble sont toujours dans un rapport de nombres entiers simples. Par exemple, l’oxygène (O) et le carbone (C) capable de réagir ensemble de plusieurs façons.
1g de C + 1.33g de O → 2.33g de CO
1g de C + 2.66g de O → 3.66g de CO2
pour former du monoxyde de carbone ou du dioxyde de carbone. Le rapport entre la masse d’oxygène est de 1 à 2, c’est la base de la stoechiométrie. Bertholet a protesté contre cette loi parce que l’une de ses expériences ont donné des résultats opposés. Cette expérience a impliqué un solide de CuO dans laquelle le rapport entre Cu et O n’ est ni constant ni un nombre entier simple. La raison en est que les solides peuvent avoir des imperfections. Fondamentalement, ces imperfections peuvent être vides ou remplacées par d’autres atomes. C’est pourquoi Bertholet a obtenu une formule de Cu1-xO au lieu de CuO.
Dalton a établi une théorie atomique :
1) Toute la matière est faite d’atomes. Les atomes sont indivisibles et indestructibles.
2) Tous les atomes d’un élément donné sont identiques (la masse et les propriétés). Les atomes d’éléments différents sont différents.
3) Les composés sont formés par une combinaison de deux ou plusieurs atomes. Il n’y a aucune formation de nouvel atome (sauf réactions nucléaires).
4)Une réaction chimique est un réarrangement des atomes.
La masse de chaque élément a été déterminé.Les premières œuvres ont été réalisées par Cannizzaro fondant son expérience sur un principe énoncé par Avogadro : Dans des conditions normales de température et de pression, des volumes identiques de gaz ont le même nombre de particules. En connaissant la proportion de carbone dans les différents gaz existant Cannizzaro a pu déterminer sa masse:
| Composé | Mass (g) | % de carbon | Mass de carbon (g) |
| Methane | 16 | 75 | 12 |
| Ethane | 30 | 80 | 24 |
| Propane | 40 | 82 | 36 |
La masse de C a été déterminée de cette façon. Le carbone a une masse de 12 unités de masse atomique(u). Consécutivement la masse d’oxygène (16) a été déterminée à partir de dioxyde de carbone (CO2) et ainsi de suite. Initialement certaines erreurs se sont produites, généralement en raison d’éléments avec une même masse. Par exemple, on sait que 2 g de H réagit avec 16 g de O pour former 18 g d’eau. Considérant la relation simple H a une masse de 1u (ce qui est exact) mais O aurait une masse de 8 u.
Nombre de moles et nombre d’ Avogadro
La mole est l’une des sept unités du Système international (unités de SI) : kg pour la masse, le mètre pour la longueur, la seconde pour le temps, Kelvin pour la température, ampère pour le courant électrique, candela pour l’intensité lumineuse et la mole pour le montant de substance. Le symbole de mole est mol.
Pour en revenir à Avogadro, l’un des nombres les plus importants dans la chimie, mais presque jamais utilisé est le nombre d’Avogadro (NA). Comme les atomes sont incassables, il ya évidemment plusieurs atomes dans 12g de C. Une mole exprime le nombre d’atomes de carbone dans 12 g de carbone.
MC = NA.mC
Cette relation est valable pour n’ importe quel élément i. Mi est la masse molaire du i, c’ est-à-dire la masse d’une mole de l’élément i. Ses unités sont g / mol (ou g mol-1). mi est la masse d’un atome de l’élément i. Dans le cas du carbone, MC = 12 g mol-1. mC étant une masse, unité NA est mol-1. La valeur de NA a été initialement déterminée par Johann Josef Loschmidt qui a calculé le nombre de particules dans un volume donné de gaz. La précision de la mesure était perfectible et il ya maintenant des expériences qui donnent des résultats plus précis que cette méthode.
NA= 6.02214129(27)×1023 mol−1
Nous-mêmes et notre environnement sont donc remplis par un nombre étonnamment grand d’atomes qui interagissent ensemble pour former la matière, de l’air, des liquides et surtout la vie. L’idée que les molécules de la vie pourraient être fabriqués n’a pas été acceptée avant le XIXe siècle ( Friedrich Wöhler)
Chapitre 2a:réactions acides-bases
Dans ce module, nous allons examiner l’un des principaux types de réaction de la chimie. Les réactions peuvent effectivement être classés en trois grandes catégories :
– réactions acides-bases
-réactions d’oxydo-réduction(rédox) .
-réactions de solubilité (de dissolution et de précipitation)
Les deux derniers types de réaction seront vus dans d’autres sections de nos leçons. Ici nous allons mettre l’accent sur les réactions acide-bases. La première étape sera d’introduire les définitions de l’acide, de base et la notion d’acidité. Nous verrons ensuite la force de différents acides et bases et nous etudieront ensuite la neutralisation d’un acide par une base.
Definitions
Un proton est un atome d’hydrogène (H) qui a perdu son électron (e-). Par conséquent un proton est seulement constitué d’un noyau de l’atome d’hydrogène qui est chargé positivement. Nous avons là à considérer que le noyau d’un atome est seulement une très petite fraction du volume d’un atome (rayon de ![]() ). En raison de sa petite taille, un proton peut diffuser partout et se déplacer dans n’ importe quel matériel jusqu’à sa neutralisation.
). En raison de sa petite taille, un proton peut diffuser partout et se déplacer dans n’ importe quel matériel jusqu’à sa neutralisation.
Plusieurs définitions ont été données à un acide et une base. Arrhenius a proposé :
-un acide est un donateur de protons
-une base est un donateur de OH-
Cette définition explique bien beaucoup de réactions, par exemple :
![]()
![]()
Toutefois, certains composés basiques ne possèdent pas de groupe OH et peuvent encore neutraliser les acides. Par exemple NH3 peut réagir avec H +, mais ne peut pas libérer de OH-. Une tentative d’explication était d’introduire NH4OH :
![]()
mais ce composé n’ existe tout simplement pas.
Brønsted and Lowry ont proposé une autre théorie :
-un acide est un donneur de protons.
-une base est un capteur de protons.
Lorsqu’une base réagit avec un acide, ils forment respectivement leurs acide et base conjugués
![]()
Consirérons un acide HA,et une base B, l’équation peut être écrite :
![]()
HA perd un proton pour former sa base conjuguée A- . La base B reçoit le proton pour former son acide conjugué HB +.
Le point intéressant avec cette théorie, est que l’acidité d’un composé dépend de la réaction dans laquelle il intervient. Il permet à certains composés d’être considérés à la fois comme un acide et une base. Par exemple H2O peut donner ou recevoir des protons.
![]()
ce type de composé est appelé amphotère. Il y a donc à la fois de l’acide et de la base dans l’eau. Toutefois lorsque nous buvons ou que nous mettons notre main dans l’eau nous ne sentons pas ces substances, pourquoi? La raison est que les trois substances (H2O, H3O+ et OH-) sont en équilibre. La réaction juste au-dessus va dans les deux sens, comme il est indiqué par la double flèche. Cependant, la réaction ne va pas dans les deux directions à la même vitesse. La constante d’équilibre pour la réaction allant de gauche à droite est Kw=10-14mol2l-2=[H3O+][OH–]. Alors que celle de la réaction allant de droite à gauche est K=1/Kw=1014mol-2l2. Cela signifie que l’équilibre est fortement orienté vers la gauche. Il n’ est donc pas fréquent qu’une molécule de H2O soit autoprotolysée et lorsque ça se produit, la réaction inverse est très rapide.
Avec Kw, nous pouvons déterminer la concentration de protons (ou H3O+) dans l’eau.
![]()
comme H3O+ et OH- sont produits au même rythme, leur concentration est égale : [H3O +] = [OH-]. Donc :
![]()
Dans l’eau pure, à tout moment, la concentration en protons est donc 10-7 M (M = moles / l). Si un acide est mis dans l’eau, la quantité de protons dans la solution augmente. Inversement, si une base est mise dans l’eau, la quantité de protons diminue. L’acidité d’une solution est alors mesurée par la concentration des protons dans la solution. Pour plus de confort l’échelle, appelée potentiel hydrogène ou pH, est diminuée du logarithme de la concentration en protons :
![]()
et va de 0 à 14 dans des solutions aqueuses. A pH = 0, la concentration des protons dans la solution est de 1M. A pH = 14 presque tous les protons sont éliminés de la solution par la base. Le pH n’est pas infinie, car il existe toujours quelques protons restants dans la solution en raison de l’équilibre. pH = 7 est un pH neutre et c’est le pH de l’eau pure. La plupart des espèces vivantes sont adaptés à ce pH neutre. D’autres se sont adaptées à des conditions basiques ou acides pour éviter la prédation ou concurrence pour les ressources.
Nous pouvons également parler de pOH pour les bases avec pOH = -log [OH-]. pOH n’est cependant généralement pas utilisé. Pour la solution aqueuse basique il est plus facile de se référer à pH = 14 pOH = 14 + log [OH-].
Lewis : acids et bases
La même année Gilbert Newton Lewis a proposé une alternative plus large pour la définition des acides et des bases : une base de Lewis est définie comme un composé qui peut donner une paire d’électrons à un acide de Lewis, un composé qui peut accepter une paire d’électrons. Considérant les mêmes notations que ci-dessus
![]()
Les deux points dans cette notation représente la paire d’électron que la base de Lewis B et le A de base conjuguée portent. Le proton est un acide de Lewis, en acceptant les paires d’électrons. Avec une telle définition les acides ne sont plus limités aux substances portant des atomes d’hydrogène. Par exemple BF3 est un acide de Lewis comme l’alésage(bore) peut accepter une paire d’ électrons.
La dissociation de H et OH
Il est à noter que dans certains cas H n’est pas dissocié comme un proton. Lorsque la liaison entre deux atomes est brisée, la paire d’électrons reste avec un atome d’une plus grande électronégativité (χ-).
Exemples:
H-Cl: χ–Cl=3.16 χ–H=2.2
Dans ce cas, et comme prévu, la paire d’électrons reste sur l’atome de chlorure, car son électronégativité est supérieure à celle de l’hydrogène.
Na-H: χ–Na=0.93 χ–H=2.2
L’hydrure de sodium est l’une des rares exceptions où l’atome d’hydrogène prend la paire d’électrons. En effet, son électronégativité est supérieur en comparaison avec Na.
Si nous regardons maintenant à la liaison OH :
χ–O=3.44 χ–H=2.2
Ce groupe, typique dans les composés basiques peut se briser pour libérer un proton. Une molécule portant un groupement OH peut alors être acide ou basique en fonction de l’atome lié à l’oxygène.
Dans NaOH par exemple l’électronégativité de Na (Na-χ = 0,93) est plus petite que celle de l’hydrogène (χ-H = 2,2), et de loin. Par conséquent, c’est la liaison entre O et Na qui rompt. Comme le O est déjà chargé négativement, la liaison OH ne sera pas divisée pour donner O2 et H +.
Au contraire, dans HClO (Cl-OH), la différence d’électronégativité entre Cl-O est plus grande que celle entre OH. En conséquence, l’acide hypochloreux se divise en ClO- et H +.
En résumé, l’acidité d’une substance dépend de la réaction dans laquelle il prend part et la présence d’un atome d’hydrogène ou un groupe OH dans la substance ne veut pas dire qu’il s’agit d’un acide ou d’une base et vice versa le fait qu’une substance est acide ou basique ne signifie pas que cette substance porte un groupe H ou OH.
Mesure du pH
Il existe différentes méthodes pour mesurer ou donner une idée du pH d’une solution.
indicateur de pH
Lorsque quelques gouttes d’indicateur de pH sont ajoutées à une solution, l’indicateur de pH de la solution donne une couleur en fonction de l’acidité de la solution. Dans une zone donnée de pH la solution sera d’une certaine couleur tandis que la couleur est différente dans une autre zone de pH. Ces zones ne sont pas spécifiquement entre 0-7 et 7-14 et dépendent de l’indicateur de pH utilisé. Le changement de couleur est due à des interactions entre les protons et les molécules de l’indicateur de pH.
Par exemple le vert de bromocrésol est jaune sous sa forme acide et bleu dans sa forme de base. Il ya une zone de transition de pH pour le vert de bromocrésol entre le pH de 3,8 à 5,4 où sa couleur est verte, la couleur de « sa forme neutre » (en fait c’ est un mélange de l’acide et de base ). La structure de vert de bromocrésol est montrée est la figure 1. La couleur de la solution ne varie pas sensiblement dans la même zone de pH mais seulement à la limite entre les deux zones. Il est expliqué par le fait que seules quelques gouttes suffisent pour obtenir une couleur visible. En outre, comme il existe des interactions entre l’indicateur de pH et les protons, le pH de la solution est affecté par la présence de l’indicateur de pH.
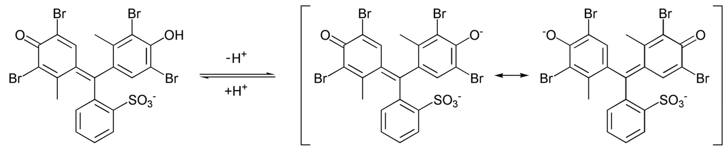
Figure 1 : Structure du vert de bromocrésol dans sa forme acide (à gauche) et basique ( au milieu et à droite). Il existe deux structures de résonance pour la forme basique. La resonance sera vu dans d’autres chapitres (chimie organique)
Notez que, à pH = 5,5 par exemple, cet indicateur est dans sa forme de base même si la solution est acide. Les indicateurs de pH sont donc utiles d’avoir une idée de l’acidité de la solution. Cependant beaucoup d’indicateurs de pH existent et sont faciles à utiliser.
Papier pH
Le papier pH est un document contenant plusieurs indicateurs de pH.

Initialement jaune sa couleur varie, en fonction du pH de la solution du rouge profond pour acides à bleu profond pour les bases. Habituellement une gouttelette de la solution est tombé sur le papier pH, ce qui lui donne sa couleur. On peut ensuite comparer la couleur du papier pH à l’échelle sur la boîte du papier pH pour déterminer le pH de la solution.
pH-mètre
Ce dispositif détermine la concentration des protons dans la solution grâce à une électrode plongée dans la solution. Il est plus précis que les deux autres méthodes mais peut avoir besoin d’étalonnage. Son fonctionnement sera vu plus tard.
Chapitre 2c : neutralisation et titration
Neutralisation:
Une réaction de neutralisation est la réaction qui a lieu entre un acide et une base formant un sel et de l’eau.
![]()
Techniquement la neutralisation n’est pas une réaction à une seule étape, en ce sens que toutes les mesures ne sont pas effectuées simultanément mais pas à pas. La première étape est la dissociation de l’acide et de la base à partir de leurs espèces conjuguées. La deuxième étape est la formation du sel à partir des espèces conjuguées et de l’eau à partir de H3O+ et de OH–. Par exemple NaOH est neutralisé par HCl pour former NaCl (sel de cuisine) et de l’eau.
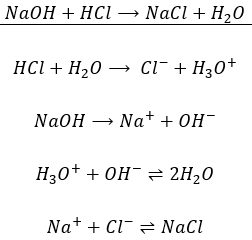
La première équation est en effet l’addition des quatre équations situées en dessous. Si un composé se trouve des deux côtés de l’équation, comme les espèces ioniques, nous ne les écrivons pas dans l’équation moyenne. Notez que toutes les molécules de Na+ et de Cl– ne réagissent pas pour donner NaCl. Il y a ici un équilibre entre les espèces en solution et la précipitation du sel. Cet équilibre doit être vu dans la section de la dissolution.
Le point de neutralisation est atteint lorsque les quantités d’acide et de la base mises en solution sont égales. Tous les réactifs sont consommés et ensuite pour cette réaction, le pH est neutre c-à-dire pH = 7.
Titrage des acides et bases fortes :
Le titrage est une méthode utilisée pour déterminer la concentration d’un composé à travers sa neutralisation. Par exemple la concentration d’une solution d’HCl peut être déterminée par l’addition d’une solution de NaOH. La concentration de la solution de titrage est connue. Comme nous sommes en présence d’un acide fort (HCl) et d’une base forte (NaOH), les 2 produits seront complètement dissociés en solution. Dans HCL La concentration en protons est initialement équivalente à la concentration en Cl-. Dans l’autre solution, la concentration en OH- et en Na+ est aussi équivalente.
Pour obtenir un pH neutre le nombre de protons na (en moles) doit être égal au nombre de OH–, nb (en moles). En d’autres termes la neutralisation ou l’équivalence, est atteinte lorsque :
![]()
Le nombre de moles d’un composé dans une solution n’est que la concentration de cette espèce multiplié par le volume de la solution :
![]()
A partir des deux relations précédentes nous pouvons trouver la concentration initiale de l’acide que nous voulions déterminer :
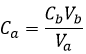
A titre d’exemple, si 20 ml de NaOH 0,01 M a été nécessaire pour neutraliser un volume de 10 ml de la solution de HCl, alors [HCl] = 0,02 M.
Au laboratoire, les titrages sont effectués comme suit :
Un ballon contenant un volume donné de la solution inconnue est placé sur un agitateur magnétique. La puce magnétique est placé dans la solution pour mélanger de façon continue pendant l’expérience. Pour pouvoir observer le processus de neutralisation deux gouttelettes d’indicateur de pH sont ajoutées à la solution. Plusieurs indicateurs peuvent servir à observer le passage à travers pH = 7. Ici, nous allons utiliser le bleu de bromothymol dont la couleur passe du jaune (pH <6,0) au bleu (pH> 7,6). A pH = 7, la couleur est le vert. Considérant l’exemple précédent notre solution est donc jaune. On peut déjà dire que la solution est acide et que son pH est inférieur à 6.
La solution de neutralisation NaOH 0,01M, dans notre cas, remplit une burette placée quelques centimètres au-dessus de l’autre solution. Il n’y a pas besoin d’ajouter un indicateur de couleur dans cette solution. Manipulant avec soin la burette NaOH est ajouté lentement à la solution acide. On peut lire le volume consommé de la base sur les graduations de la burette. Lorsque le pH se rapproche de 6 on peut voir les gouttelettes de base tourne au bleu tout en se mélangeant dans la solution. Ne connaissant pas du tout la concentration de l’acide il est commode d’effectuer une expérience rapide pour déterminer un volume approximatif pour la neutralisation et d’effectuer une seconde expérience, en allant lentement à l’approche de ce volume. Typiquement la couleur passe du jaune au vert ou directement au bleu à la chute d’une gouttelette. La précision de cette expérience est donc limitée à la précision de la burette. En général le volume d’une gouttelette est la moitié d’une graduation.
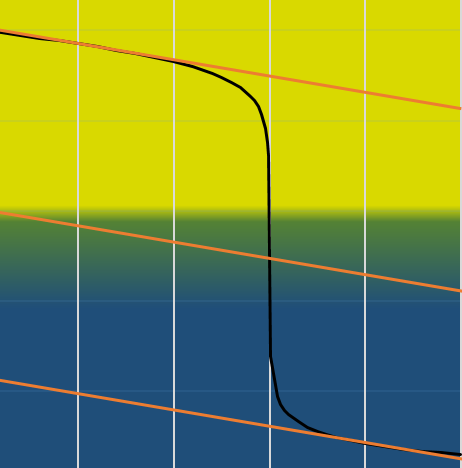
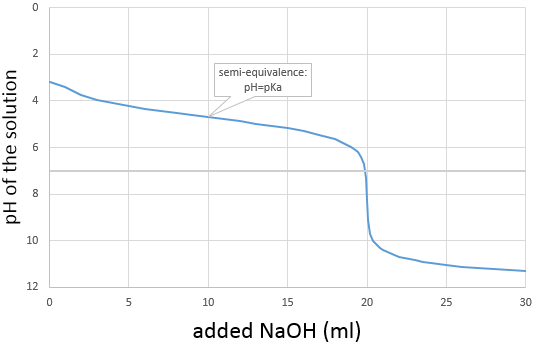
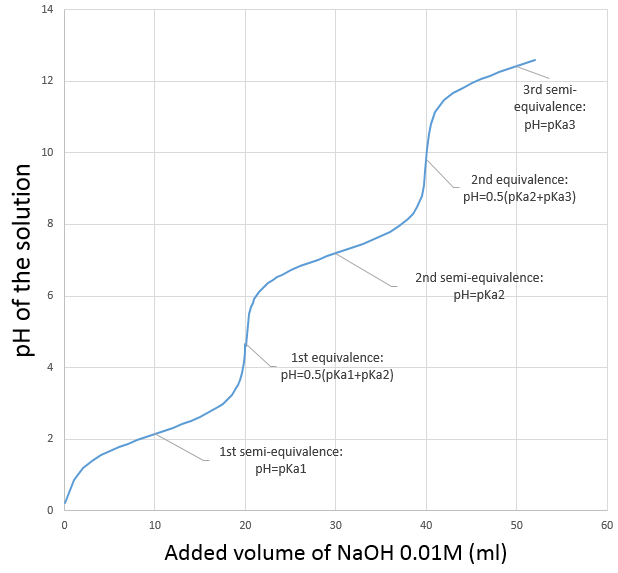
La courbe de titrage de cet exemple est représenté ce dessous.
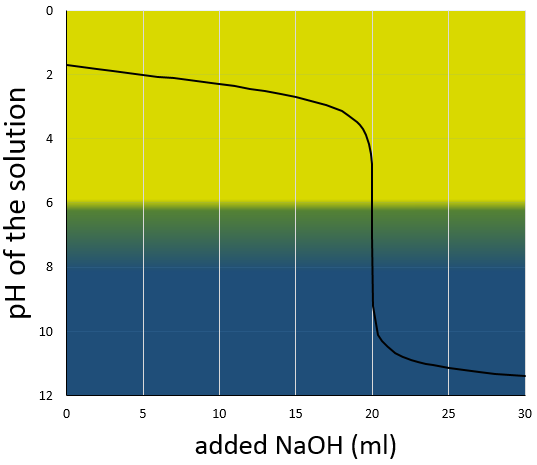
Rappelons que le pH est le logarithme de la concentration des protons alors que l’addition de la base est linéaire et que le volume de solution augmente avec l’addition de la base (ne pas oublier ce point lors du choix du volume de la fiole). Comme nous pouvons le voir la variation de pH se concentre principalement au voisinage du point de neutralisation. À 20 ml, pH = 7 mais à 19.95ml (un gouttelette de moins environ, pour une burette de 50ml), pH = 4,78 et au 20.05ml, pH = 9,22.
Nous pouvons également trouver le point d’équivalence en traçant les tangentes de la courbe dans la région acide et dans la région basique. Ces deux tangentes sont parallèles. Lorsque nous traçons la ligne équidistante à ces deux lignes, elle rencontre le point d’équivalence.
Titration des acides et bases faibles :
Le titrage d’un acide faible est effectué en utilisant une base forte et suit le même principe qu’en haut. Prenenons comme exemple le titrage de l’acide acétique la réaction est :
![]()
Cette réaction est complète. Avant le titrage le pH de la solution d’acide acétique est simplement donnée par la relation d’un acide faible que nous avons vu dans la section précédente :
![]()
En ce qui concerne les acides forts, l’équivalence est atteinte lorsque :
![]()
Cependant le pH à l’équivalence n’ est pas neutre mais basique. En effet, les espèces conjuguées d’une base/acide forts sont inertes mais les espèces conjugués d’un acide / base faibles sont eux-même une base / acide faibles : pKa + pKb = 14. Tout l’acide acétique et NaOH sont consommés mais acétate a été produite et c’ est une base faible.
Avant l’équivalence, le pH dépend de la quantité d’acide faible et de sa base conjuguée :
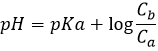
Ce mélange entre un acide faible et sa base conjuguée est appelé une solution tampon car l’addition d’une base forte ou d’acide ne modifie pas sensiblement le pH de la solution. Les solutions tampons sont très importantes pour les espèces vivantes qui les aident à résister à des variations brutales de l’environnement. Un exemple de solution tampon est l’estomac. Peu importe ce que nous mangeons ou buvons son pH n’est presque pas affecté de sorte que le travail peut être fait. Dans notre corps les enzymes sont efficaces dans une région donnée du pH et pour les garder efficaces le pH doit être maintenu dans ces zones par des solutions tampons.
La semi-équivalence est le moment où il y a autant de CH3COOH que CH3COO–(Ca=Cb). À ce stade pH=pKa. Pour atteindre le point semi-équivalant le volume supplémentaire de base est la moitié du volume pour obtenir l’équivalence. A l’équivalence le pH est donné par la quantité d’acétate dans la solution (formule pour une base faible). Cette quantité est égale à celle de NaOH ajouté à la solution.
Après le point d’équivalence le pH est donné par la quantité de NaOH dans la solution.
Le titrage d’un polyacide :
Considérons un polyacide HnA (un exemple concret sera donné plus loin) avec assez de différents pKa, la neutralisation des différentes formes de l’acide sont successives: la OH– d’abord va neutraliser les protons libérés par HnA puis les protons libérés par Hn-1A– etc…..
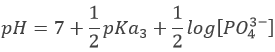
Le pH initial de la solution est le pH deHnA. Les concentrations des acides suivants sont négligeables. HnA peut être un acide fort ou un acide faible.
Au premier point d’équivalence Hn-1A– est la principale espèce en solution. C’ est une espèce amphotère c’est à dire qu’il peut accepter ou donner des protons. Le pH est ainsi :
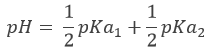
Au point de semi-équivalence suivant,[Hn-1A–]=[Hn-2A2-] et nous sommes dans une solution tampon. Rappelez-vous que le pH dans des solutions tampons est
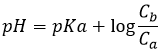
Le pH est donc pH =pKa2. Notez que si l’acide initial est un acide faible la même chose est vraie pour le premier point de semi-équivalence, à savoir pH =pKa1. Il est intéressant de noter que, pour ces valeurs particulières, le pH ne dépend pas de la concentration.
Prenons le cas de H3PO4 à titre d’exemple. Ses pKa sont très différents les uns des autres
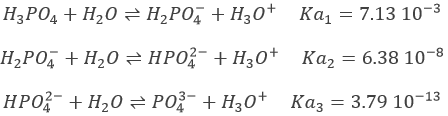
Les neutralisations sont successives et nous pouvons ainsi trouver les points spécifiques (semi-équivalences et équivalences) déterminés ci-dessus.
H3PO4 est un acide faible. Le pH initial de la solution est donc :
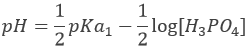
Avant l’équivalence, H3PO4 et H2PO4– sont en solution. Cette solution tampon a un pH de :
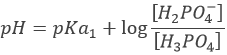
Avec pH = pKa1, lorsque [H3PO4] = [H2PO4–], à la mi-équivalence.
Lors de la première équivalence, H2PO4– est la principale espèce en solution. C’ est une espèce amphotère et le pH sera par conséquent:
Après la première équivalence, H2PO4– et HPO42- sont en solution. C’ est encore une solution tampon.
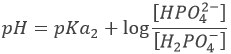
Avec pH = pKa2 lorsque [H2PO4–] = [ HPO42-], soit à la deuxième demi-équivalence. Au deuxième point d’équivalence HPO42- est la principale espèce en solution et est amphotère.
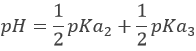
Après cette équivalence, la solution est à nouveau une solution tampon.
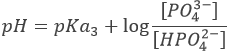
Avec pH = pKa3 lorsque [HPO42-] = [PO43-] c’ est à dire à la troisième demi-équivalence. Lors de la troisième équivalence PO43- est la principale espèce en solution. Ce n’ est pas une espèce amphotère mais une base faible. Le pH doit être
Cependant, Ka3 est très proche de Kw . Les protons libérés par l’eau sont en concurrence avec des protons de HPO42- et la prédiction n’est plus possible. Pour calculer le pH nous devons revenir à la composition complète de la solution puis résoudre l’équation. Ce ne sera pas fait ici.
Exercices:
1) Quelle est la couleur du bleu de bromothymol dans une solution de 20 ml de NaOH 0,005 M ? Si l’on ajoute 10 ml, 20 ml ou 30 ml de HCl 0,005 M?
2) Quelle est la couleur du bleu de bromothymol dans une solution de 20 ml de H3PO4 0,1 M ? Si l’on ajoute 10 ml, 20 ml ou 30 ml de NaOH 0,1 M?
Réponses:
1) 0 ml : bleu (pH = 11,7), 10 ml : bleu (pH = 11,22), 20 ml : vert (pH = 7), 30 ml : jaune (pH = 3).
2) 0 ml : jaune (pH = 1,57), 10 ml : jaune (pH = 2,147 pKa1), 20 ml : jaune (pH = 4,67), 30 ml : vert (pH = pKa2 = 7,2).
Article traduit de l’anglais ( cf site de BORZUYA en anglais )
Chapitre 2b: la force des acides et bases
La définition générale d’un acide est donc un composé libérant des protons. Toutefois, tous les acides n’ont pas la même force ou acidité. Nous pouvons définir deux types d’acides et bases : des acides et bases forts et des acides et bases faibles. Pour plus de simplicité, nous allons nous concentrer sur les acides dans cette leçon, mais le principe est identique pour les bases.
Les acides forts
Les acides forts se dissocient totalement en solution. Cela signifie que toute molécule d’acide mise dans l’eau permettra de libérer un proton et d’acidifier la solution. Par exemple, HCl est un acide fort.
![]()
Si une mole de HCL est mise dans l’eau, tout le HCL se dissocie en solution et nous ne pouvons plus trouver de HCL mais plutôt une mole de Cl- et une mole de H3O + . Pour ce genre de réaction la flèche séparant les réactifs des produits est une simple flèche allant de gauche à droite. Comme la réaction va seulement dans un sens, le potentiel hydrogène ou pH, peut donc tout simplement être trouvé avec la quantité de HCL mise en solution. pH = -log [H3O +] et dès que la réaction est terminée, la quantité de H3O + dans la solution est égale à la quantité de HCL mise en solution. La concentration en protons est donc égale à la concentration de HCL dans la solution avant la réaction [H3O +] = [HCL] 0.
Par exemple, si 0,1 mole de HCL est mise dans l’eau pour obtenir un volume total de 1 litre, [H3O +] = 0,1 mol / l, le pH sera = 1. Au laboratoire, en général, l’acide est déjà en solution avec une forte concentration (par exemple 6 M) et doit être dilué à la concentration désirée pour l’expérience. N’oubliez pas que des précautions doivent être prises lorsque vous manipulez des acides et des bases, en particulier avec les produits concentrés. Utilisez une poire et la pipette et non votre bouche. Une autre règle «sainte» est que «On ne baptise pas un acide », ce qui signifie que pour diluer un acide il faut ajouter l’acide dans l’eau et non l’eau dans l’acide. La raison en est que la dilution d’un acide est fortement exothermique et des gouttelettes d’acide peuvent être éjectées hors du récipient.
L’effet de la dilution sur le pH est simple. Si une solution a un pH = 2 ([H3O +] = 0.01mol / l) elle est diluée 10 fois par rapport à un PH=1(le pH augmente par une unité alors que la concentration diminue de 10 fois) car c’est une échelle logarithmique. et pH = 3 ([H3O +] = 0.001mol / l), etc. Pour les bases, une dilution diminue le pH de la solution à la neutralité (pH = 7). C’ est en effet la concentration de OH qui est affecté dans ce cas. Comme pH = -log [H3O +] = 14 + log [OH-].
En outre, une grande dilution de l’acide n’ entraîne pas une solution basique. La dilution par 100 d’une solution de pH = 6 ne donne pas une solution de pH = 8 mais environ pH = 7. Dans ce cas, l’eau est l’espèce principale définissant le pH. La concentration des protons provenant de l’acide devient négligeable par rapport à la concentration de protons libérés par l’eau.
Pour être considéré comme un acide fort, la constante de dissociation de l’acide doit être suffisamment grande pour transformer toutes les molécules de H2O de la solution en ions H3O +. Formellement les acides forts ont un pKa <-1,74. Nous avons vu que l’eau a une constante de dissociation de Kw=……………… .La constante de dissociation de l’acide est noté Ka. De la même façon que le pH est -log de la concentration en protons, pKa = -log Ka. Par exemple HBr a un pKa de -8,7. La limite de pKa <-1,74 est simplement la concentration de l’eau :
Dans 1l d’eau il y a 1 kg de H2O. La masse molaire de H2O étant égale à 18.01528g, la concentration de l’eau pure est [H2O] = 55,5084. -log De cette concentration est 1,74.
En résumé, pour pouvoir ioniser toutes les molécules d’eau de la solution qui est la condition pour être considéré comme un acide fort, l’acide doit avoir un pKa <-1,74. Certains acides couramment utilisés sont généralement considérés comme des acides forts mais ne répondent pas à cette condition ( ces acides ont un pka comprises entre 0> pKa> -1,74 ) car ils se dissolvent complètement dans la solution diluée. Ce sont les acides presque forts.
Parmi les acides forts, nous pouvons trouver de l’acide chlorhydrique (HCl, l’acide pratiquement fort), de l’acide sulfurique (H2SO4), l’acide nitrique (HNO3, l’acide pratiquement fort), l’acide iodhydrique (HI), l’acide percloric (HClO4), l’acide bromhydrique (HBr) et beaucoup d’autres.
Des exemples de bases fortes : hydroxyde de sodium (NaOH), l’hydroxyde de potassium (KOH), hydroxyde de calcium (Ca (OH) 2), …
La base conjuguée d’acides forts sont des bases très faibles et sont inertes comme base. En effet, la basicité de la base conjuguée d’un acide (et inversement) est liée à la Ka de l’acide. La relation est Ka.Kb = Kw = 10-14. Imaginez une seconde que la base conjuguée réagit avec l’eau. Si l’on ajoute les réactions de l’acide et de sa base conjuguée, nous obtenons la autoprotolyse d’eau:
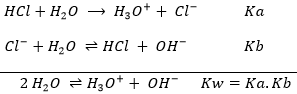
HCl a un Ka = ![]() et le Kb de Cl- est donc Kb =
et le Kb de Cl- est donc Kb = ![]() .
.
Acides faibles :
Tous les acides ne se dissolvent pas complètement dans l’eau. Les acides avec pKa> 0 sont considérés comme des acides faibles. Étant donné que toutes les molécules d’acide ne se dissolvent pas, il existe un équilibre entre les molécules dissociées et les molécules non dissociées de l’acide.
![]()
L’équilibre est représenté par les deux flèches entre les réactifs et les produits.
Un exemple d’acide faible est l’acide acétique (CH3COOH).
![]()
En solution, il est donc un mélange de ces quatre molécules. Le pH est toujours déterminé par la quantité de protons en solution. Comment Pouvons-nous déterminer cela dans le cas des acides faibles ?
La constante de réaction de cette réaction est
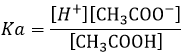
Jetons un oeil à la concentration des espèces avant et après la réaction
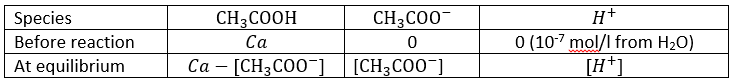
Une partie de la concentration initiale de l’acide (Ca) a réagi. La quantité de protons et de la base conjuguée (CH3COO-) produit après la réaction sont égales.
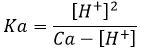
En général, Ca >> [H +], conduisant à la relation suivante.
![]()
Le pH de la solution peut donc être trouvé à partir de la concentration initiale de l’acide mis en solution et de son Ka :
![]() .
.
Pour bases, la relation est similaire:
![]() .
.
Ces relations peuvent aussi s’ écrire :
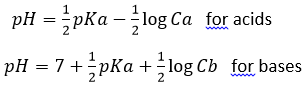
A partir de ces équations, on peut voir directement pourquoi ce type d’acide est faible en ce qui concerne les acides forts : Pour obtenir une augmentation du pH de 1, un acide fort est dilué par 10 ,tandis qu’un acide faible doit être dilué par 100.
Contrairement aux acides forts, les bases conjuguées d’acides faibles sont actives en tant que base dans la solution. Par exemple, la base conjuguée d’un acide faible avec un Ka = 10-4 a un Kb =10-10 .
Facteurs influants sur l’acidité :
Électronégativité : L’électronégativité se réfère à la capacité des atomes de garder leurs électrons ainsi que les électrons des liaisons qu’ils partagent, près de leur noyau. Pour deux atomes liés ayant la même électronégativité, les électrons qui composent le lien ne sont pas statiques mais ils passent un temps égal à chaque extrémité de la liaison (statistiquement). Pour les deux atomes liés ayant une électronégativité différente, les électrons passent plus de temps dans le voisinage de l’atome d’électronégativité plus grande. Cela génère une séparation des charges, un dipôle avec une charge partielle négative (noté δ-) sur l’élément électronégatif et une charge positive partielle (noté δ +) sur l’élément électropositif. Dans les acides, H possède une charge positive partielle en fonction de l’électronégativité de l’atome auquel il est lié .

Cette charge partielle positive stimule la dissociation de H et augmente ainsi l’acidité de la molécule. Jetons un oeil à la table de Mendeleïev, se déplaçant de gauche à droite sur une même ligne les éléments du tableau périodique deviennent plus électronégatifs (à l’exclusion des gaz nobles) et la force de l’acide formé par ces éléments et les atomes d’hydrogène augmente en conséquence.
Les éléments électronégatifs qui ne sont pas directement liés à l’atome d’hydrogène peuvent également tirer des électrons de l’hydrogène. L’effet est beaucoup plus petit mais ne devrait pas être négligé.
Rayon : les électrons de liaison des atomes plus volumineux sont situés plus loin de leur noyau que ceux de petits atomes. Du fait de cette distance ces électrons sont moins énergétiques : il existe une interaction plus faible avec le noyau et la charge du noyau est partiellement protégée par les électrons des couches internes. En conséquence la liaison est plus facilement cassée pour libérer un proton si l’atome se liant à l’hydrogène est grand. La séquence d’acidité pour les acides halogènes montre cela clairement. HF (acide Hydrofluorous) est un acide faible (pKa = 3,2) et qui est moins acide que HCl, HBr ou HI, même si son électronégativité est supérieure à la leur car elle est beaucoup plus petite (d’un facteur 2 à 3). La liaison entre le fluor et l’hydrogène est donc plus fort parce que les électrons sont à proximité du noyau. HI est le plus grand de la séquence et est aussi l’acide halogène le plus acide avec un pKa de -9,3> pKaHBr (-8,7)> pKaHCl (-6,3)) >> pKaHF (3,2). En descendant une colonne sur la table de Mendeleïev la taille des éléments augmente et deviennt moins électronégative. L’effet de la taille a tendance à dominer la variation de l’électronégativité et de l’acidité des composés portant des atomes d’ hydrogène.
Polyacides :
Plus tôt nous avons mentionné l’acide sulfurique, H2SO4. Cet acide a deux protons disponibles.
Plus haut, nous avons mentionné l’acide sulfurique, H2SO4; cet acide a deux protons disponibles.
![]()
![]()
H2SO4 est un acide fort (pKa1=-3). Lorsque l’acide sulfurique est mis en solution un premier proton est libéré et il ne devrait pas rester en solution H2SO4. D’autre part HSO4– est un acide faible (pKa2=1.9) et il ne sera pas totalement dissocié dans l’eau. Pour déterminer le pH, nous pouvons procéder comme nous l’ avons fait pour un acide faible:

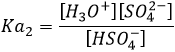 En appliquant les règels mathématiques on peut voir que [HSO4–] = 2Ca- [H3O +] et on peut écrire:
En appliquant les règels mathématiques on peut voir que [HSO4–] = 2Ca- [H3O +] et on peut écrire:
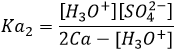
et a partir de cela nous obtenons une équation du second degré :
![]()
qui doit maintenant être résolu pour obtenir :
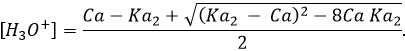
Le pH peut encore être calculé à partir de la constante de dissociation et de la concentration initiale de l’acide mis en solution même si la solution est un peu plus difficile.
Produits amphotériques :
sont des produits qui présentent deux caractéristiques acides et basiques. HCO3– est un exemple de produit amphotérique. Comme l’acide sulfurique H2CO3 est un polyacide. Cependant l’acide carbonique est un acide faible et il existe donc un équilibre impliquant HCO3– en tant que base conjuguée de H2CO3.
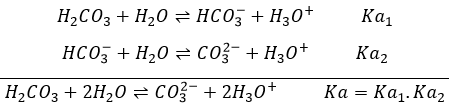
L’eau est également une espèce amphotère comme elle peut libérer ou accepter un proton. Il existe un pH particulier auquel l‘espèce amphotère a le même effet que d’une base comme d’un acide. Ce pH est appelé le pH isoélectrique ou PI. Pour H2O dont le pH isoélectrique est de 7 mais il peut être déterminée à partir de la valeur de Ka :
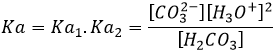
Considérant que [H2CO3]=[ CO32-] à ce pH,
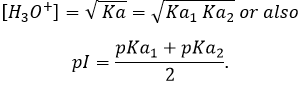
L’acide aminé est une autre espèce amphotère. Ce type de molécule porte un groupe acide et un groupe basique. La figure 1 montre la structure d’un acide aminé : forme acide (à gauche), neutre (au milieu) et la forme de base (à droite). Le groupe acide de l’acide aminé est le groupe COOH. Son atome d’hydrogène peut être libéré afin d’obtenir une charge négative sur l’oxygène. Cette charge est stabilisé par résonance par le groupe COO. Le groupe aminé sur la gauche de la molécule joue le rôle de la base. L’azote possède une paire d’électrons disponibles pour accepter un proton.
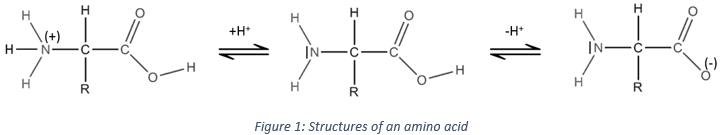
Cette propriété amphotère de l’acide aminé est utilisé expérimentalement pour séparer des acides aminés entre eux. En raison de leur structure particulière (R varie en fonction de l’acide aminé), chaque acide aminé a un pH isoélectrique différent. Les molécules sont placées sur un gel contenant un gradient de pH, à l’intérieur d’un champ électrique. Tant que l’acide aminé n’ est pas dans sa forme neutre il est attiré par une électrode placée à une extrémité du gel. Chaque acide aminé cessera donc de se déplacer à un endroit différent sur le gel. Par exemple Alanine (R =CH3) a un pI = 6 alors pi = 5,48 pour la phénylalanine (R=CH2C6H5).
Exercices:
Vous pouvez trouver ici quelques exercices pour appliquer la théorie expliquée dans cette section et, éventuellement, les sections relatives à son sujet. La plupart des questions sont simple à répondre, mais certaines peuvent nécessiter une calculatrice ou être un peu plus compliquées. Les réponses sont données en dessous.
1) Quel est le pH d’une solution de HCl 0,5 M?
2) Comment puis-je proceder expérimentalement pour obtenir 100 ml de HCl 0,05 M?
3) Quel est le pH de cette solution?
4) Si je mets une goutte de cette solution sur un morceau de papier de pH, quelle couleur le papier prendra-t-il?
5) HClO ou (Cl-O-H) est-ce un acide ou une base?
6) Quel est le pH d’une solution à 0,025 M de HClO (pKa = 7,497) ?
7) Si cette solution est diluée par 10 quel sera son pH ? et si dilué par un autre 10 ?
8) Quel est le pH d’une solution de NaOH 0,01 M ? 0,01 M de NH3 (Kb = 1,8 × 10−5) ? A l’équilibre, combien NH3 reste en solution?
Réponses:
1) pH = 0,3: HCl est un acide fort et se dissocie complètement en solution pH = -log [H3O +] = – log [HCl] = 0,3
2) Même si dans ce cas, l’acide n’est pas très concentré il a encore un pH élevé et des précautions doivent être prises pour manipuler une telle solution. Ici, nous avons juste besoin de faire une dilution de la solution acide de 10 fois. Pour obtenir 100 ml de solution diluée, nous aurons besoin d’une pipette de 10ml et sa poire, et une fiole jaugée de 100 ml. On ne baptise pas un acide. Le flacon est donc d’abord rempli avec un volume d’eau (50 ml par exemple) et 10 ml de la solution acide est ajoutée en utilisant la pipette et sa poire. Mélanger la solution et ajouter de l’eau jusqu’ à l’obtention du volume exacte. Mélangez une fois de plus.
3) pH = 1,3: comme la solution initiale a été diluée par 10, pH augmente de 1. Pas besoin de calculer ici.
4) Rouge foncé
5) HClO est un acide : il a été expliquée en détail dans la section précédente : dans HClO (Cl-OH) la différence d’électronégativité entre Cl-O est plus grand que celui entre OH. L’acide hypochloreux se divise en ClO- et H +.
χ–Cl=3.16, χ–H=2.2, χ–O=3.44
6) pH = 4,55. L’acide hypochloreux est un acide faible. La formule de pH est alors :
![]() 7) PH = 5,05 pour une dilution par 10 et 5,55 pour une dilution par 100.
7) PH = 5,05 pour une dilution par 10 et 5,55 pour une dilution par 100.
8) NaOH: pH = 12, NH3: pH = 10,63, [NH3] = 0.009576M. Nous pouvons ici simplement utiliser la formule des bases faibles pour le NH3 (rappelez-vous que le pKa + pKb = pke), mais nous aurons besoin des détails de la troisième partie de la question développons le problème :
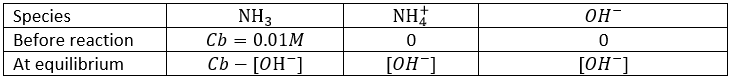
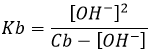
Considérant que Cb >> [OH-] et nous allons voir que cette approximation est correcte, nous trouvons :
![]()
Donc, à partir du 0,01 M de NH3 qui a été mis en solution seulement ~ 4% dissocier. La concentration résiduelle de NH3 est [NH3] = 0.009576M. pH est trouvé en utilisant pH = 14 + pOH = 10,63.
Chapitre 3: Chimie organique, la structure des alcanes
La chimie organique est la chimie du carbone et de ses composés. Le carbone est un élément de le tableau de Mendeleïev parmi beaucoup d’autres, alors pourquoi y a t-il une section complète de la chimie liée à cet élément particulier ? Le carbone a une valence de 4 et peut donc se lier avec quatre autres éléments de la classification périodique. Jusque là, il n’y a rien d’extraordinaire. Toutefois, lorsque les atomes de la chimie inorganique ne se lient ensemble que pour former de petites molécules, des molécules à base de carbone peuvent former de longues chaînes stables avec une riche variété de groupes fonctionnels.
La chimie organique est appelée ainsi parce que le carbone est le constituant essentiel des espèces vivantes : les protéines, l’ADN, les lipides, les sucres ou les graisses sont quelques exemples de composés organiques. Ces composés ont une structure de carbone portant des groupes fonctionnels leur permettant d’interagir ensemble, devenir non seulement une simple addition d’atomes, mais pour former un macrosystème de fonctionnement où chaque molécule a un rôle spécifique à maintenir un corps vivant stable.
La chimie organique est donc très importante dans la science de la vie mais n’ est pas limitée à cela. On trouve le carbone dans le plastique , l’huile, le dentifrice, les shampooings, les vêtements, des déodorants, etc. tous ces produits et bien d’autres sont des composés organiques.
Pour nous aventurer dans ce vaste monde qui est la chimie organique, nous allons d’abord discuter sur les alcanes, leurs structures, leur appellation scientifiques , et leur représentation spaciale. Plus tard, nous allons présenter les différents groupes fonctionnels que nous pouvons trouver sur les composés organiques et enfin comment les composés organiques réagissent, comment nous pouvons les produire ou les modifier.
Alcanes
Les alcanes sont des composés constitués uniquement d’atomes de carbone et d’hydrogène. L’hydrogène a une valence de 1, ce qui signifie qu’il ne peut faire qu’une seul liaison avec un autre atome. Un seul carbone va donc se lier à 4 atomes d’hydrogène pour former une espèce neutre CH4. Cette molécule est appelée méthane et dans des conditions normales est un gaz. Les liaisons sont des liaisons covalentes. Le carbone est légèrement plus électronégatif que l’hydrogène, mais il n’ est pas important à ce moment de la leçon. N’oubliez pas que CH4 ne peut pas se dissocier pour donner un proton et n’est donc pas un acide.
Il ya plusieurs façons de représenter cette molécule. La représentation entièrement développée est la suivante :
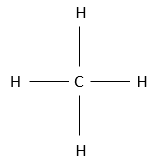
Dans cette représentation du méthane, toutes les liaisons sont représentés par des lignes reliant les atomes. Tous les atomes sont présentés indépendamment ainsi. Cette représentation est en deux dimensions mais en réalité les positions des atomes d’hydrogène en 3D ne sont pas dans un plan unique. La structure moléculaire avec la plus faible énergie est la structure dans laquelle les atomes d’hydrogène sont les plus séparés (éloignés) les uns des autres. En effet, les atomes d’hydrogène ont un volume donné et se repoussent mutuellement. Pour obtenir cette structure un angle de 109,5° sépare les liaisons. Elle conduit à une structure tétraédrique.
La plupart du temps il est inutile de montrer la structure compliquée de grosses molécules. Cela ne ferait que rendre plus difficile de voir les informations importantes de la molécule. Cependant il est parfois important de savoir dans quelle direction va une liaison particulière. Dans ce cas les lignes, représentant les liaisons, prennent des formes différentes en fonction de leur orientation. Dans le plan de la page, les liaisons sont encore représentées par une ligne simple. Deux autres cas sont possibles : Les liaisons peuvent aller dans le sens du lecteur ou dans le sens opposé. Les liaisons allant vers le lecteur sont représentées par un triangle noir une crête est reliée à l’atome sur le plan de la page et les deux autres points sont reliés à l’atome situé en dehors du plan. De cette façon le triangle ressemble à une ligne qui va de l’atome situé sur le plan à l’atome situé plus près du lecteur.
Pour les liaisons qui vont dans la direction opposée, les atomes sont reliés par des traits. Pour le méthane il donne la représentation 3D qui suit :
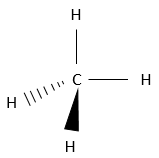
L’alcane possédant une structure de 2 atomes de carbone est l‘éthane. C’est un gaz comme le méthane. Pour former l’ossature de la structure de l’éthane, les deux atomes de carbone se lient ensemble par une liaison covalente. Il est évident qu’ils ont la même électronégativité. Leurs quatre électrons, sont donc utilisés par chaque carbone pour se lier avec l’autre. En outre 6 hydrogènes complètent la structure. Comme dans la chimie inorganique, la règle de l’octet est respectée : pour être stable un carbone doit avoir 8 électrons (un octet) autour d’elle (contraignants ou non) : ses quatre électrons et quatre électrons des autres atomes avec lesquels il partage une liaison. Chaque carbone de l’éthane est donc lié à un atome de carbone et trois atomes d’hydrogène . C’ est la seule structure possible pour un tel composé. En aucun cas un carbone porterait 5 hydrogènes et l’autre carbone 1 ou plus.
La structure pleinement représentée de l’éthane est donc
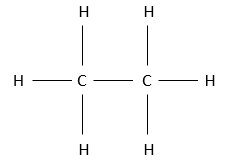
Si nous voulons la représenter en 3 D, ce serait :
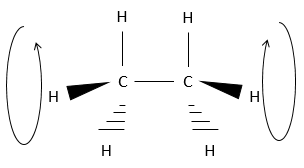
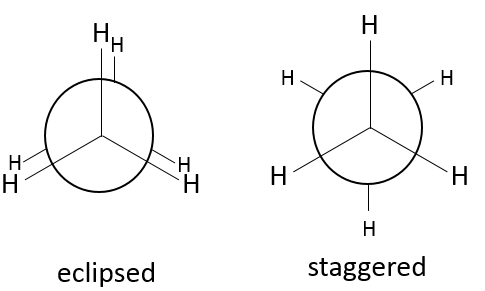
Cependant il faut signaler que les atomes peuvent tourner dans l’axe d’une liaison simple. Les 6 hydrogène se tournent ainsi en cercle autour de l’axe formé par les deux atomes de carbone, comme indiqué ci dessus.
Chaque hydrogène a un volume donné et sent les atomes dans son voisinage (encombrement stérique). Lors de la rotation la distance entre les hydrogènes du même carbone reste constante. Toutefois la distance avec l’hydrogène le plus proche porté par les autres carbones change.
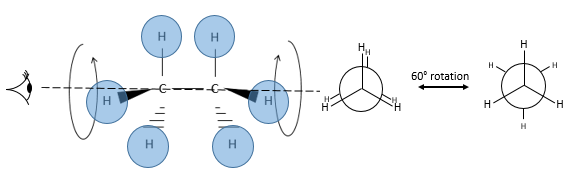
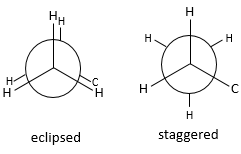
Les positions relatives des substituants peuvent être montrées par la projection de Newman. La molécule est observée le long de son axe C-C. Le premier C (carbone proximal) est représenté par un cercle et à partir du centre de ceci trois lignes sont dessinées vers la périphérie. Ces lignes sont les liaisons de ce carbone. Le second carbone (carbone distal) est caché par le premier, mais une partie de la liaison est visible.
Fixant les hydrogènes du carbone proximal, seulement les hydrogènes du carbone distal peuvent se déplacer. Deux cas peuvent être observés :
– Les hydrogènes du carbone distal et proximal sont sur les mêmes points, et sont » éclipsés »
– Les hydrogènes du carbone distal et proximal ne sont pas sur les mêmes points et sont en quinconce « staggered »
Un maximum d’énergie est atteinte dans la conformation de l’éclipse parce que la répulsion entre les hydrogènes est maximale dans cette conformation. Une rotation de 60° par rapport à cette conformation conduit à un minimum d’énergie car les hydrogènes sont le plus loin possible les uns des autres. Une molécule qui doit maintenir l’hydrogène (ou des substituants) en forme éclipsée a une énergie supérieure par rapport à une molécule de même composition avec des hydrogères sous forme étalée (staggered). La différence d’énergie ici n’ est pas très importante et la rotation est efficace. Pendant la rotation, la molécule passe plus de temps dans la conformation décalée (plus petite énergie). Si les substituants sont sur la molécule, l’encombrement stérique augmente avec le rayon du substituant. Dans certains cas la rotation peut être bloquée par la présence de substituants volumineux.
La semi représentation de l’ éthane est:
![]()
Dans cette représentation les atomes de carbone sont regroupés avec les atomes qu’ils portent. Les liaisons entre C et H ne sont donc pas montrées dans cette représentation. Si un atome d’hydrogène a été remplacé par un atome de chlore, par exemple, la semi représentation serait :
![]()
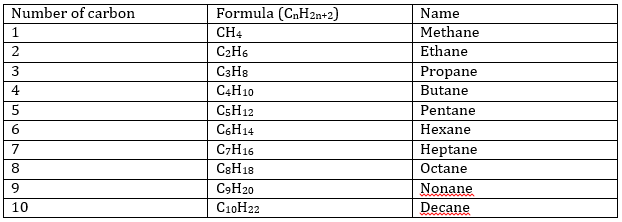
L’ajout d’un troisième atome de carbone à la chaîne donne le C3H8 (propane), qui est encore un alcane linéaire. La structure en triangle où chaque atome de carbone se lie à deux autres carbones existe mais n’est pas stable. Vous avez peut-être remarqué que la formule des alcanes a un modèle général : CnH2n+2. Pour chaque atome de carbone ajouté à la chaine deux atomes d’hydrogène doivent être ajoutés.
![]()
Il ya deux façons d’ajouter un quatrième carbone pour obtenir une molécule de butane. La chaîne peut être étendue par le côté ou par son milieu. Lorsque la chaîne est linéaire, on ajoute n- avant le nom du composé. n-butane est donc
![]()
Si la chaîne est prolongée par son milieu, nous nommons ce composé isobutane
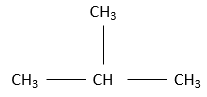
Le préfixe iso est utilisé seulement pour quelques composés dans lesquels un carbone terminal peut porter deux bornes CH3. n-butane et l’isobutane ont la même formule C4H10 mais n’ ont pas la même structure. Ce type de composé est appelé un isomère. Plus le nombre de carbone augmente dans un alcane, plus le nombre d’isomères augmente.
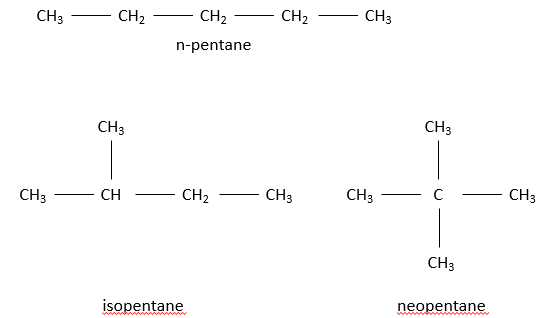
Pour une chaîne de 5 carbones le cinquième carbone peut être ajouté à une extrémité du n-butane pour obtenir un n-pentane et s’il est ajouté à n’ importe quelle extrémité de l’isobutan on obtiendra l‘isopentane par contre s’il est ajouté sur le CH de l’isobutane on obtiendra le néopentane.
Les noms des alcanes :
Une autre représentation des molécules organiques est la représentation de l’ossature. Dans cette représentation les atomes de carbone et les atomes d’hydrogène ne sont pas représentés. Les liaisons entre les atomes de carbone sont toujours représentées par des lignes complètes et sont reliés entre eux avec un angle à la position des atomes de carbone. Sans angle nous ne pourrions pas différencier une chaîne de six atomes de carbone d’une chaîne de 7 atomes de carbone. En règle générale l’angle est d’environ 120° de sorte que si un carbone est lié à trois autres espèces (autres que H) chaque liaison soit à égale distance. Si un atome de carbone est lié à quatre espèces l’angle est de 90°.
Par exemple, les pentanes montrés ci-dessous sont représentés :
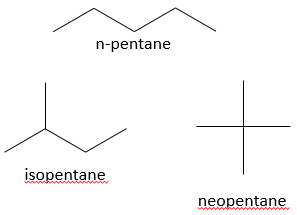
Cette structure de type ossature est la représentation qui est habituellement utilisée. Seuls les informations importantes sont affichées. Le nombre d’atome d’hydrogène sur les différents atomes de carbone de l’ossature sont déterminés par le nombre de possibilités de liaison que le carbone a. Il est donc inutile de le montrer. En outre cette représentation est plus rapide à écrire et prend moins d’espace.
Il y a une méthode donnée pour nommer les composés organiques. Un alcane en tant que groupe fonctionnel a le même nom sauf que le -ane est remplacé par -yl. Par exemple, l’isobutane est aussi appelé méthylpropane parce qu’un groupe méthyle est fixé à une chaîne linéaire de 3 atomes de carbone, soit une chaîne de propane.
C4H9Cl est un Chlorobutane. Avec ce nom, nous connaissons les composants du composé, mais pas sa structure complète. La connectivité dans le butane et entre le butane et le chlore n’est pas connue.
Les règles pour nommer un composé sont :
– la plus longue chaîne est la principale toutefois si un groupe fonctionnel est sur une chaîne, la chaîne principale doit le porter.
exemples :
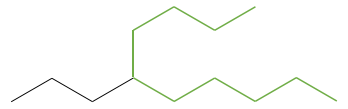 is a decane
is a decane
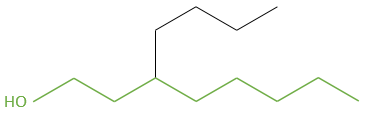
is an octanol
– marquer un numéro à chaque carbone à partir d’un côté de la chaîne principale à l’autre. Le carbone portant un groupe fonctionnel qui est le plus proche d’une extrémité doit avoir le plus petit nombre.
toujours avec les mêmes exemples :
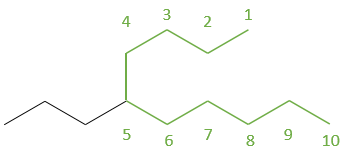
Le carbone portant la petite chaîne de la gauche a le plus petit nombre posssible de cette façon.
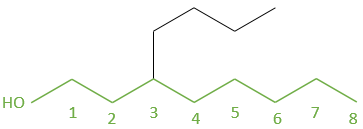
Nous énumérons de gauche à droite en raison de la présence du groupe OH sur la gauche. Ensuite nous nommons le composé en écrivant d’abord les groupes sur la chaîne principale avec leur numéro comme préfixe dans l’ordre alphabétique, suivie par la chaîne principale avec son groupe.
les exemples sont ainsi nommés
5-propyldecane
3-butyloctan-1-ol
Si plusieurs groupes fonctionnels identiques sont sur différents atomes de carbone, les préfixes sont séparés par un………, et leur nombre est indiqué par bi, tri, …
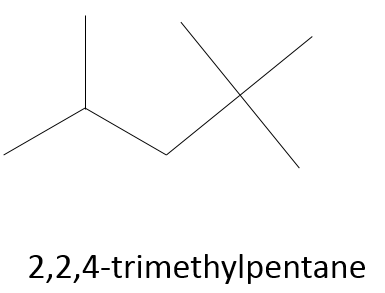
ex : l’iso-octane est le 2,2,4-triméthylpentane ce qui signifie qu’un total de trois groupes méthyle sont sur une chaîne principale de 5 carbones. Deux sont des méthyles sur le carbone N°2 et l’autre sur le N°4. C’est le 2,2,4-triméthylpentane et non 2,4,4-triméthylpentane parce que nous privilégions le carbone portant plusieurs groupes
Halogenoalcanes :
Les halogénoalcanes sont des alcanes portant un ou plusieurs halogènes. Il est simplement dit mais les halogenoalcanes ne sont pas si facilement fait. Ils sont fabriqués à partir d’un alcane dihalogéné et un par une réaction radicalaire au cours de laquelle un proton doit être retiré de l’alcane. Cette étape de la réaction n’ est pas favorable mais peut être faite par chauffage lourd (300° pour le chlorométhane). En outre la position de l’halogène n’est pas complètement résolue. Le proton supprimé pendant la réaction est plus facilemet retiré d’un carbone de la chaîne à une extrémité. Mais la température élevée rend les deux positions possibles (la distribution dépend de la température).
Les halogènes ont une électronégativité supérieure à C et elles génèrent un dipôle de C à X. Un carbone portant un halogène est donc pauvre en électrons et par conséquent peut être ciblé par des réactifs riches en électrons. La réactivité des halogénoalcanes sera vue dans une autre section.
Les molécules chirales :
Nous avons déjà vu que, pour une formule donnée, plusieurs molécules différentes peuvent exister. Lorsque la connectivité diffère, ce sont des isomères de constitution.
Ex : butane et méthylpropane, éthanol et méthoxyméthane.
Même sur un seul carbone, la connectivité peut changer. Les stéréoisomères sont des isomères de même connectivité, mais avec le positionnement spatial différent. Le bromochlorofluorométhane a deux formes de stéréoisomères.
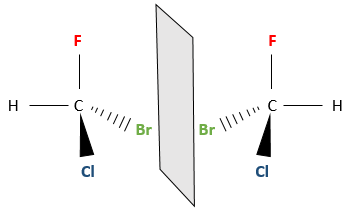
Ces deux molécules sont des images en miroir l’une de l’autre. On dit qu’elles sont chirales si la molécule et son image en miroir ne se superposent. Ces stéréo-isomères particuliers sont appelés énantiomères.
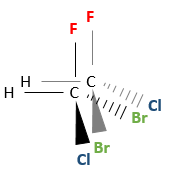
Une bonne manière d’expliquer la chiralité est de regarder nos mains. La main gauche est l’image en miroir de la main droite (et vice versa). Cependant nous ne pouvons pas les superposer.
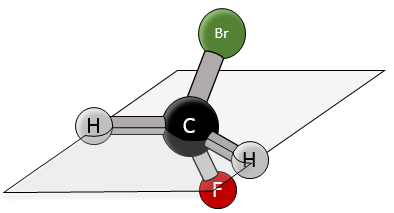
La chiralité est liée à l’atome de carbone qui porte plusieurs groupes différents c’ est un stéréocentre. Les stéréocentres sont souvent signalés par un astérisque. Si un plan de symétrie existe pour la molécule, alors cette molécule est achirale (><chiral) et la molécule et son image en miroir peuvent en quelque sorte se superposer. Par exemple parce que le Bromofluorométhane est achiral un plan de symétrie peut être tiré, en passant par les deux atomes d’hydrogène.
Notre corps est capable de distinguer l’une de l’autre des énantiomères. Pour certains médicaments un énantiomère est actif tandis que l’autre ne fera absolument rien ou sera moins efficace. Dans certains cas, il est donc très important d’être capable de produire sélectivement un énantiomère et pas l’autre. Les industries pharmaceutiques ont investi ces méthodes. Si la réaction n’ est pas énantiosélective, la productivité chute immédiatement de 50%. L’activité optique des énantiomères diffère également et est un bon moyen de savoir quels énantiomères ont été produites.
l’activité optique
L’activité optique d’un composé est son influence sur un faisceau de lumière polarisée dans un plan. Lorsque la lumière filtrée à osciller uniquement dans un plan, passe à travers un échantillon d’un composé optiquement actif, le faisceau est mis en rotation d’un angle donné.
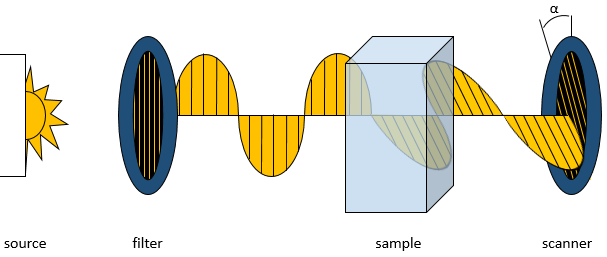
L’angle de rotation dépend des molécules dans l’échantillon, de leur concentration et de la longueur de la cellule d’échantillonnage. Chacun de ces effets sont linéaires et la modification de l’angle de la lumière est donnée par la formule
![]()
L’intérêt de ce phénomène est que les énantiomères n’ ont pas la même activité optique. La modification de l’angle absolu est identique, mais la direction dans laquelle la lumière est mis en rotation ne l’est pas. Un énantiomère dévie la lumière vers la droite et l’autre énantiomère dévie la lumière par exactement le même angle, mais vers la gauche. Les énantiomères sont respectivement définis comme dextrogyre et lévogyre et asont notés avec un (+) ou un (-).
L’activité optique d’un énantiomère est fixée, à une température donnée t et pour une lumière de longueur d’onde λ donnée. En connaissant la valeur de l’activité optique, il est possible de déterminer la quantité de chaque énantiomère dans un mélange racémique. Un mélange racémique est une solution contenant les deux énantiomères, pas nécessairement dans la même quantité (nb : une définition plus globale d’un mélange racémique est le mélange de plusieurs produits possibles d’une seule réaction). Dans un tel mélange, toutes les espèces dévient la lumière par leur effet normal.
Si les deux énantiomères sont en quantités égales dans la solution l’échantillon sera optiquement inactif, l’effet sur un énantiomère étant contrebalancé par l’effet de son image spéculaire (ie l’autre énantiomère). Si les quantités ne sont pas égales l’échantillon est optiquement actif et les quantités relatives des énantiomères peut être déterminée. L’excès énantiomérique est la différence de proportion des deux énantiomères est pratiquement et la proportion des énantiomères qui ont un effet sur la lumière. Par exemple si le rapport entre les énantiomères était de 3: 1 (75% de l’un (disons le dextrogyre) et 25% de l’autre énantiomère) l’excès énantiomérique est de 50%. En effet à partir de 75% de l’énantiomère dextrogyre l’effet de 25% est contrebalancé par l’énantiomère lévogyre présent dans la solution. Seulement 50% de l’énantiomère (+) dévie le faisceau lumineux. Si l’énantiomère pur aurait tourner le plan de lumière de 26°, un excès de 50% entantiomeric la lumière tourne de 13° (50% de 26 °).
![]()
Nom des énantiomères :
Nous devons trouver une façon de nommer différemment les énantiomères. R ou C seront précédés par le nom de la molécule et nous allons maintenant voir comment attribuer quelle lettre à quel énantiomère. C’est malheureux mais il n’y a pas de corrélation claire entre le pouvoir rotatoire d’un énantiomère et sa structure. Une autre méthode a dû être trouvée.La première étape consiste à donner une priorité à chaque groupe fixé à un stéréocentre.
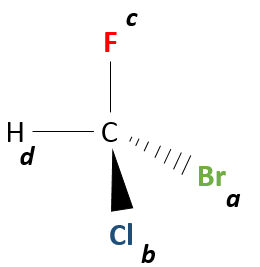
La priorité est donnée à l’atome qui a la masse la plus élevée en relation avec le stéréocentre. Appelons ces groupes a, b, c et d par une diminution de la priorité (A a la priorité sur B, B sur C, et C sur D). Si deux atomes ont le même poids (deux atomes de carbone sont liés au stéréocentre par exemple), nous regardons les atomes qu’ils portent et, encore une fois, la priorité va au carbone portant l’atome de poids atomique le plus grand. Si un groupe méthyle et un groupe éthyle sont liés au stéréocentre, l’éthyle a la priorité. Les deux groupes sont liés au stéréocentre par un atome de carbone. Nous étudions maintenant les atomes sur ces carbones. Le méthyl a 3H etl’éthyle a 2H et une C. Comme C est plus lourd que H, l’éthyle a la priorité sur le méthyle.
Rappelez-vous que c’est le poids de l’atome lié au stéorocentre qui compte, pas le poids de l’ensemble du groupe. Un groupe -OH a en effet la priorité sur un groupe éthyle parce que O (poids atomique = 16) est plus grand que C (12), même si les poids des groupes sont respectivement 17 et 29 unités de masse atomique. Notez que nous parlons de la masse des éléments. Les isotopes peuvent ainsi créer des stéréocentres dans les molécules.
Ensuite nous regardons la molécule comme si le groupe avec la plus faible priorité (D) était derrière le carbone. En général le groupe avec la plus petite priorité est un atome d’hydrogène. Cet atome (ou groupe) n’est pas représenté dans le reste du procédé.

Ainsi, en regardant au stéréocentre de cette façon, nous ne voyons que trois liaisons liant le stéréocentre avec les trois groupes de priorité (A, B, C).
Maintenant nous voulons déterminer dans quel sens faire tourner pour aller de la plus haute priorité (A) à la plus basse (C), en passant par B il peut être utile de placer A en haut de la représentation.
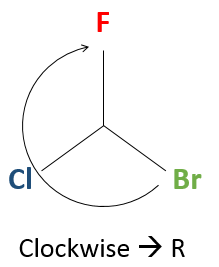
Si nous devons aller dans le sens horaire, c’ est l’énantiomère R. Et si c’est dans le sens antihoraire nous sommes en présence de l’énantiomère S.
Une deuxième méthode existe ce qui donne les mêmes résultats en utilisant la projection de Fisher.
Au lieu de placer un groupe derrière le stéréocentre on met deux groupes horizontaux et vers le lecteur par une rotation du stéréocentre. Les deux autres groupes sont sur l’axe vertical et à l’opposé du lecteur.
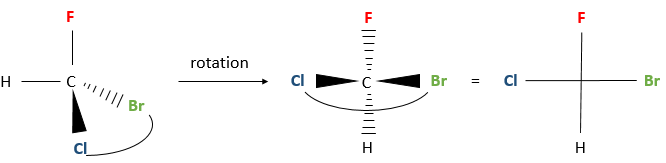
La rotation du stéréocentre est effectuée par « saisir » une paire ou des substituants et de les placer en face de la molécule. Faire attention à placer les groupes dans la direction horizontale du lecteur et les verticales dans la direction opposée. Sinon un énantiomère R est S et inversement.
Une fois que la rotation s’ effectue, afin de déterminer la configuration correcte, le groupe de priorité la plus basse doit être placé sur 12 heures de position de la représentation Fisher. Ensuite la configuration est déterminée de la même manière que pour la projection de Newman. Si le groupe de priorité la plus basse n’était pas en position haute après la rotation, ne vous inquiétez pas, nous pouvons effectuer des permutations entre les substituants voisins. L’exécution d’une permutation modifie la conformation de l’énantiomère R à S et inversement. Exécution de deux permutations laisse l’énantiomère identiques.
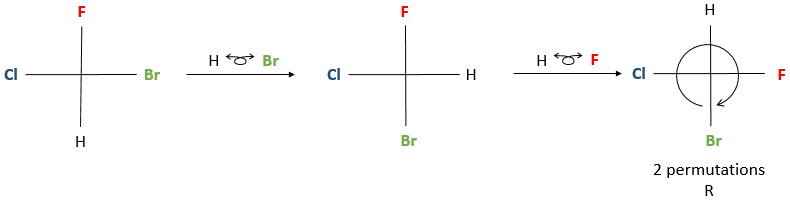
Si un nombre pair de permutations (y compris zéro) ont été faites pour mettre le groupe de priorité la plus basse dans la position de 12 heures, nous pouvons déterminer directement la configuration de l’énantiomère. Si un nombre impair de permutation a été fait alors nous avons deux choix: soit nous déterminons la configuration actuelle, sachant que la configuration correcte est l’autre, ou nous faisons une permutation supplémentaire et ensuite nous déterminons la configuration.
diastéréoisomères
Plusieurs stéréocentres peuvent être présents sur une seule molécule. La configuration de chaque stéréocentre (R ou S) est déterminée de façon indépendante. Si deux stéréocentres sont sur une molécule plusieurs configurations sont possibles : RR, SS, RS et SR.
Par exemple le 2-bromo-3-chlorobutane a quatre stéréoisomères.
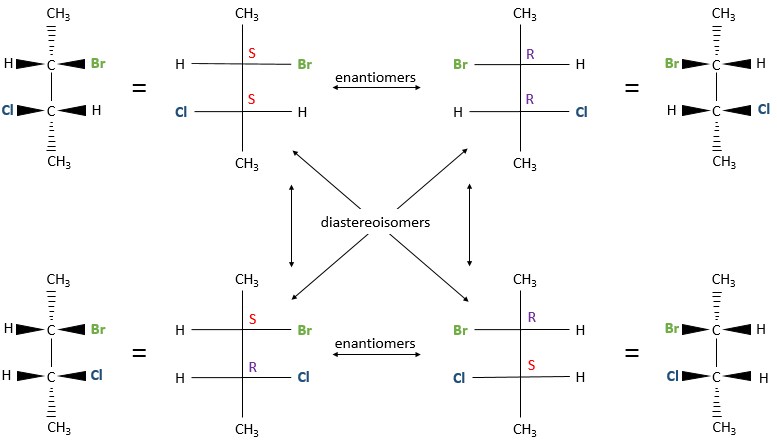
Toutes ces conformations ne sont pas des images en miroir l’une de l’autre. Certains sont stéréoisomères énantiomères mais certains ne sont pas. Les stéréoisomères qui ne sont pas l’image miroir de l’autre sont appelés diastéréoisomère.
Exercices :
Dessiner les structures d’ossature des isomères de C8H18. Combien d’isomères avez-vous trouvé?
Nommez les molécules suivantes :

3. Dessinez la molécule suivante :
3,6,10-triméthyldodecane
5-éthyl-2-méthyloctane
2,6-diméthyle-4,5-dipropylnonane
4- (1-méthyléthyl) heptane
Est-ce le nom correct ? Si non, corrigez.
2,5-diméthyle-4,6-dipropylnonane
3-éthyle-7-méthyloctane
2,5-diméthyle-4-éthyldecane
4- (1-méthyléthyle) -5-propyldecane
Dessinez les projections Newman des liaisons CC de cette molécule et précisez s’ils sont éclipsés ou décalés
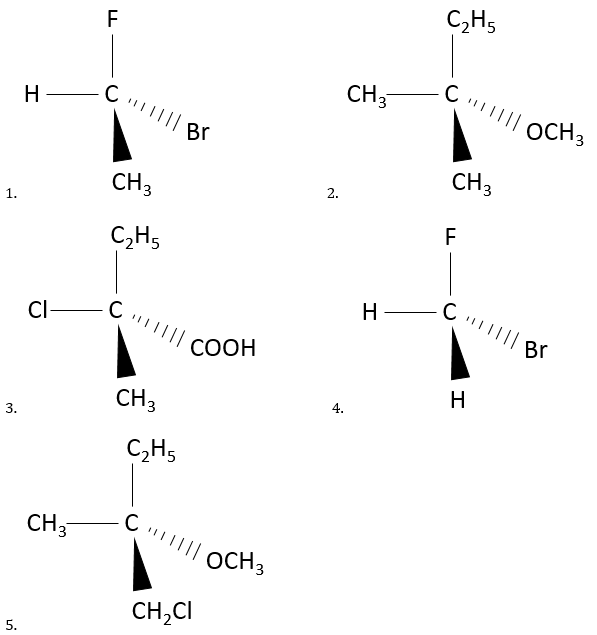
6. chiral ou achiral ? Si chiral, indiquez s’ils sont R ou S.
Réponses
1. Il ya 23 isomères de constitution pour C8H18. Une bonne façon de les trouver tous est de commencer par la plus longue chaîne principale et diminuer sa longueur étape par étape.
2. Noms :
2,1: n-hexane
2,2: Néopentane 2,2 ou 2,2-diméthylepropane
2,3: 4-éthyle-7-méthyledécane. Ce n’est pas le 4-méthyle-7-éthyledécane car les substituants sont placés dans l’ordre alphabétique s’ils sont à la même distance d’une extrémité de la chaîne
2,4 : 6-propyle-3-méthyledécane (de la chaîne principale de haut en bas à droite)
2.5: 5- (1-méthylepropyle) décane
2,6 : 4-éthyle-4-méthyloctane
3. Noms et esquisse:
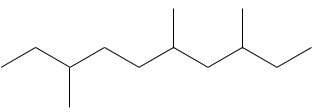
3.1: 3,6,10-triméthyledodécane
3.2 : 5-éthyle-2-méthyloctane
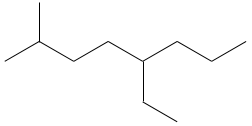
3.3: 2,6-diméthyle-4,5-dipropylnonane
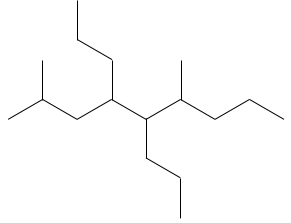
3.4: 4- (1-méthyléthyle)heptane
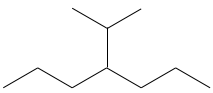
4. correcte ou non?
2,5-diméthyl-4,6-dipropylnonane: correct
Incorrect:: 7-méthyloctane-3-éthyl le substituant méthyle est plus proche d’une extrémité de l’éthyle. Le nom correct est 6-éthyl-2-méthyloctane
2,5-diméthyl-4-ethyldecane: incorrecte: substituants doivent être nommée dans l’ordre alphabétique. Le nom correct est 4-éthyl-2,5-dimethyldecane
4- (1-méthyléthyl) -5-propyldecane: correct
5. projections Newman allant de gauche à droite
6. Chiralité:
6,1 chirale: R
6,2 Achirale
6,3 chirale: S
6,4 Achirale
6,5 chirale: S
( cet article a été-traduit de l’anglais cf site en aglais de BORZUYA university )
Chapitre 3 c: les cycloalcanes
Cycloalkanes
Un cycloalcane est, comme son nom l’indique, une chaîne alcane cyclique. Chaque carbone de la chaîne est lié à (au moins) deux atomes de carbone et deux atomes d’hydrogène. La formule générale est donc CnH2n et le nom du composé est le même nom que l’alcane correspondant avec le préfixe cyclo.
Cyclopropane
Le plus petit cycle « cyclopropane est fait de trois atomes de carbone. Chaque carbone est lié aux deux autres avec une forme de triangle.
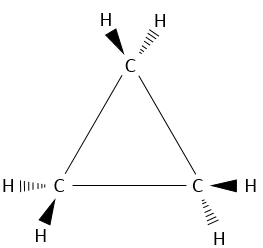
Cela veut dire que les atomes de carbone sont dans le même plan et que l’angle entre les liaisons est de 60°, cet angle est loin de l’angle normal entre les liaisons des alcanes. Rappelez-vous que les carbones ont une structure tétraédrique avec un angle de 109,5° entre chaque liaison. Pour être cyclique, il existe une déformation de la structure de carbone et une tension est maintenue dans la forme de cycle. Il est possible de déterminer l’importance de cette tension à partir de l’énergie de combustion ΔH°comb du cycloalcane.
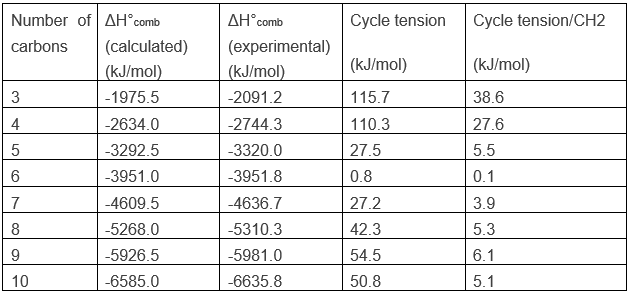
Pour un alcane linéaire la chaleur de la combustion augmente d’environ 658.5kJ/mole lorsque la longueur de la chaîne est augmentée d’une unité. On peut en conclure que le ΔH°comb moyenne d’un CH2 est 658.5kJ/mol. L’application de cela au cyclopropane, C3H6, donne un ΔH°comb calculé de -1975.5kJ/mol. Cependant, lorsque nous effectuons expérimentalement la combustion, nous constatons que AH°comb est égale à -2091.2kJ/mol.
Les cyclopropanes libèrent ainsi plus de chaleur que ce que nous pouvions espérer. La différence, 115.7kJ/mol (38.6kJ / mol / CH2), vient de la tension du cycle, c’est-à-dire que la molécule nécessite plus d’énergie pour cette forme de liaison. En fait les orbitales du carbone ne sont pas bien alignées, mais l’angle entre les orbitales est 104°.
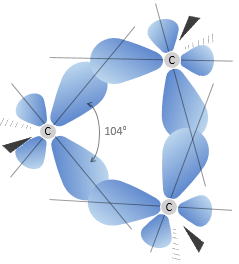
En conséquence, les liaisons sont faibles et le cyclopropane n’est pas très stable. Il est en effet facilement transformé par hydrogénation catalytique.
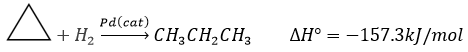
Enfin, la position des atomes d’hydrogène est défavorable. Ayons un rappel rapide des conformations éclipsées et décalées pour les hydrogènes dans de l’éthane (C2H6). L’hydrogène peut tourner autour de l’axe formé par la liaison CC. Chaque hydrogène a un volume donné et sent les atomes dans son voisinage (encombrement stérique). Lors de la rotation, pour une H donnée, la distance avec les hydrogènes les plus proches portés par des autres carbones changent.
Sur la projection Newman :
-les hydrogènes sont sur les mêmes points et sont considérés éclipsés (eclipsed)
– ou ils ne sont pas sur les mêmes points, c’est ce qu’on appelle en quinconce (staggered)
Un maximum d’énergie est atteinte dans la conformation de l’éclipse parce que la répulsion entre les atomes d’hydrogène est maximale dans de cette conformation. Pour maintenir ses atomes d’hydrogène (ou ses substituants) sous cette forme, beaucoup d’énergie est nécessaire . Dans le cyclopropane tous les atomes d’hydrogène sont éclipsés. La différence d’énergie entre les formes éclipsées et décalées peut être importante comme nous le verrons pour les cycles de plus de trois atomes de carbone.
Cyclobutane:
La combustion du cyclobutane, dont les angles sont de 90° ~, produit un excès de 110.3 kJ/mol (27.6 kJ/mol / CH2) du à la tension de cycle. Il est inférieur à la flexion, car le cyclopropane forcé est plus petite que dans le cyclobutane dans le cyclopropane. La tension de ces deux cycles sont très importants. Pour de plus grands cycles, la tension diminue de manière significative et est à son minimum pour un cycle de 6 atomes de carbone, le cyclohexane.
Les cycles avec plus de 3 atomes de carbone ne sont pas plan. Dans le cyclobutane l’angle entre le quatrième carbone de la molécule, en dehors du plan et le plan formé par les trois autres atomes de carbone est de 26°.
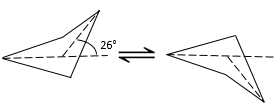
L’angle entre les atomes de carbone est 88,5° il est légèrement inférieur à un carré (90°) dans un seul plan. Alors, pourquoi le cyclobutane n’a pas une forme plane ? Il devrait en effet diminuer la tension du cycle. Toutefois dans la forme plane les 8 atomes d’hydrogène seraient éclipsés. Les 26° dans la structure du cyclobutane permettent que les atomes d’hydrogène puissent adopter la forme non éclipsée. La petite différence d’angle est ainsi compensée par l’amélioration du positionnement des atomes d’hydrogène. Il faut souligner que cette forme (avec 26 degré) peut exister sous 2 représentations spaciales et la structure du cyclobutane oscille entre les deux : l’atome de carbone en dehors du plan se déplace d’un côté du plan à l’autre, ces deux conformations sont équivalentes en énergie. En regardant la figure suivante, les 4 H sont représentés :
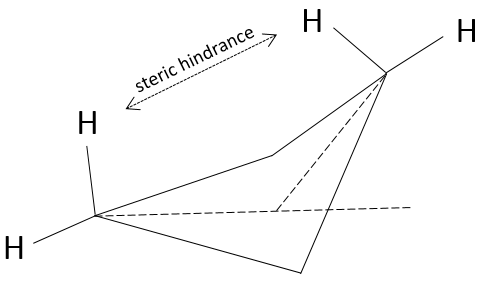
Deux d’entre eux (en remontant) sont assez proches les uns des autres tandis que les deux autres sont éloignés. Après l’oscillation les rôles sont inversés et les deux hydrogènes ont en moyenne le même encombrement stérique appelé également la tension trans-annulaire dans ce cas.
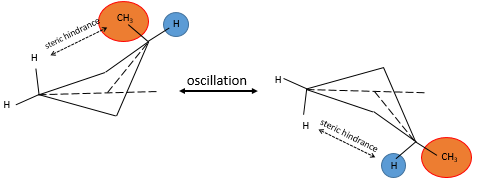
Les cycles peuvent ainsi osciller entre plusieurs conformations lorsque les conformations ont une énergie potentielle similaire et si la barrière énergétique entre différentes conformations est assez petite. Dans le cas de la cyclobutane l’encombrement est très faible mais si l’un de ces 4 hydrogènes était un substituant les deux conformères ne seraient plus équivalentes. En effet la molécule va se placer dans la conformation la plus favorable c.à.d la situation dans laquelle le substituant volumineux n’est pas affecté par la tension transannulaire. La proportion des conformères n’est plus 50:50.
Cyclopentane
L’effet défavorable de l’hydrogène dans des conformations écliptiques est clair dans le pentane. Dans un pentagone régulier, l’angle est de 108°. C’est presque l’angle normal pour un carbone tétraédrique (109,5°). Cependant, les l’hydrogènes vont éclipser les uns les autres. En fait aucan cycloalcane à part le cyclopropane, n’ est plane. Deux conformations (chacune avec deux conformères) sont possibles : l’enveloppe et le semi-chaise.
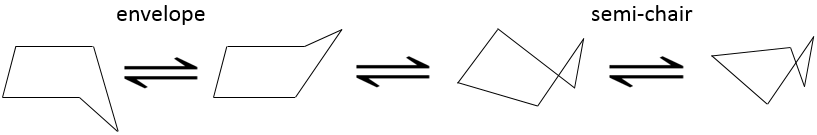
Dans la conformation de l’enveloppe 4 atomes de carbone sont dans le même plan avec un angle de 104,4°. Dans l’autre conformation (semi-chaise), les angles sont plus petits mais l’effet éclipsant est aussi plus petit. Les deux conformations ont une énergie potentielle très proche et les barrières pour basculer entre les deux formes sont facilement franchies, le cyclopentane oscille alors rapidement entre ses conformères.
cyclohexane
Le cas du cyclohexane est particulier. Quand on regarde la chaleur de combustion de cette espèce la valeur expérimentale est inférieure de (0,8) kJ/ mol par rapport à celle calculée sur la base du nombre de CH2 dans la molécule, le cyclohexane est le cycloalcane le plus stable.
Deux conformations existent mais l’une est plus stable que l’autre.
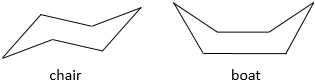
La conformation la plus stable est la conformation chaise.
Dans cette conformation deux paires d’atomes de carbone sont dans le même plan et les deux derniers atomes de carbone sont de chaque côté du plan. Cette position est appelée chaise : les quatre atomes de carbone sur le plan font le siège, un plan de trois carbone rend le dos de la chaise et l’autre fait le repose-pied.
L’angle entre les carbones est de 111,4°, soit près de 109,5° d’un carbone tétraédrique normal et tous les atomes d’hydrogène sont dans une conformation en quinconce.
Cette structure est donc très stable. Deux types d’hydrogène peuvent être distingués : ceux de la position axiale et ceux de la position équatoriale. 6 liaisons CH sont parallèles à l’axe de la molécule (axe passant au milieu de la molécule) ce sont les hydrogènes axiales. Les 6 autres liaisons sont presque perpendiculaires et sont appelées équatoriales.
cyclohexane1
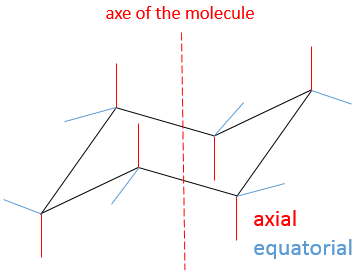
Si nous inversons la structure de la chaise (le dos de la chaise <-> le repose-pieds), les hydrogènes équatoriaux deviennent axiaux et vice versa.
Pour inverser la chaise, le cyclohexane doit passer par la conformation de bateau, moins stable par 28.9kJ/mol (la barrière énergétique est de 45.2kJ/ mol). Dans cette conformation les deux atomes de carbone qui étaient hors du plan sont maintenant sur le même côté de celui-ci. Cela ne génère pas seulement un encombrement stérique mais les hydrogènes sur les quatre atomes de carbone, sur le plan, éclipsent les uns les autres. Ce qui explique la différence de l’énergie potentielle entre la forme de bateau et les formes de chaise.
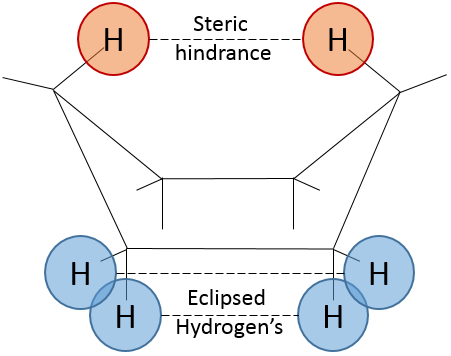
En réalité cette conformation n’est qu’un état de transition. Une forme plus stable est la conformation de bateau-croisé, presque identique, mais avec la réduction de la tension transannulaire. La conformation bateau est donc l’état de transition entre les deux conformations bateau-croisés.
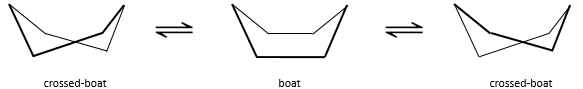
Nous pouvons résumer les conformations comme suit :
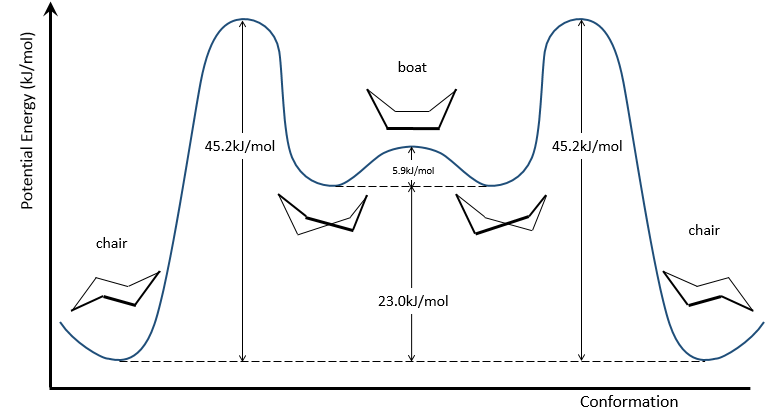
le cyclohexane dans les conformations de bateau n’existe que dans des proportions très faibles par rapport à la conformation chaise. Nous allons donc nous concentrer uniquement sur la conformation chaise pour nos autres analyses.
La présence de substituants sur le cyclohexane
Les positions d’un substituant sur le cyclohexane ne sont pas équivalentes :
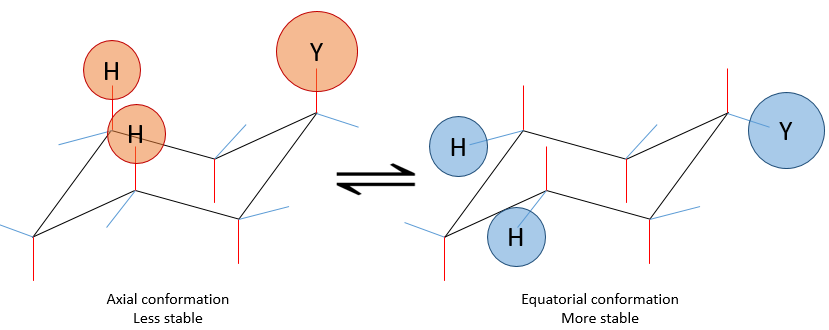
si un substituant est placé sur une position axiale l’encombrement stérique est supérieur à une position équatoriale. En effet, les substituants équatoriaux sont plus espacés que ceux axiaux qui vont dans la même direction. Ceci est appelé le 1,3-diaxial interaction. La position équatoriale pour un substituant est alors plus stable que celle axiale et une conformation de la molécule est favorisée. Par exemple, si le substituant est un groupe méthyle, la différence d’énergie entre les conformations est 7.1kJ/mol, ce qui conduit à une proportion de 95 : 5 (équatorial/axial). Les substituants plus volumineux entraînent une augmentation de la proportion de conformères.
La projection de Newman peut aider à visualiser ce phénomène. Dans le schemas ci-dessous les deux projections de NEWMAN sont représentées.
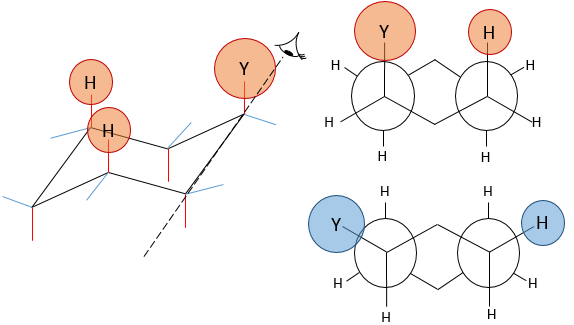
Plusieurs substituants peuvent être liés à un cyclohexane. L’influence sur la stabilité de chacun est, pour la plupart des substituants, un additif simple. Dans le cas de deux substituants on sera placé dans une position équatoriale de toute façon car il nécessite moins d’énergie que sur une position équatoriale (comme nous venons de le voir dans le cas d’un seul substituant). Ce substituant est le plus grand pour minimiser l’encombrement stérique. Le deuxième, plus petit, est sur une position axiale ou équatoriale, en fonction de la connectivité de la molécule. Si les deux groupes sont en positions équatoriales, l’équilibre est plus favorisé à l’égard de cette conformation. Si le second substituant est sur une position axiale, l’équilibre se déplace vers la composition 50:50, les effets des substituants agissent contre l’un de l’autre.
Voyons cela à travers quelques exemples. Comme expliqué précédemment la présence d’un méthyle sur le cyclohexane favorise la conformation équatoriale de 7,1 kJ/mol. La présence d’un deuxième groupe de méthyle sur un endroit équatorial va augmenter la proportion de la conformation équatoriale du même montant (7.1kJ/mol). L’avantage énergétique total de la conformation équatoriale atteint 14.2kJ/mole et la proportion des augmentations de conformères équatoriaux en outre vers 99: 1. Cette conformation est également notée trans parce que les groupes font en sens inverse. Le nom de cette molécule est en effet trans -1,4- diméthylcyclohexane.
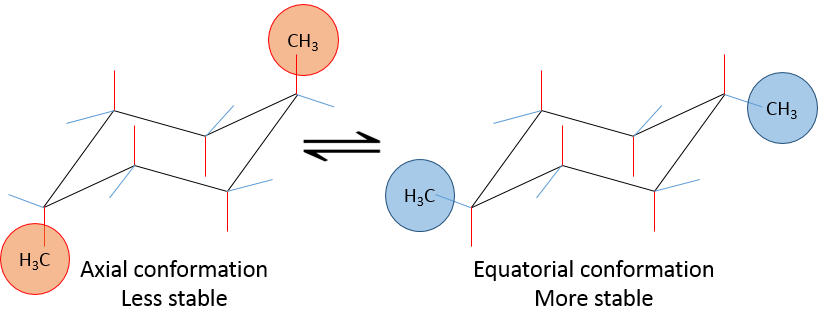
Si le second groupe méthyle était sur une position axiale les deux conformères seraient équivalents (méthyle axial devient équatorial et vice versa). L’avantage énergétique totale de l’un des conformère est en effet 7.1kJ / mol-7.1kJ / mol = 0 kJ / mol.
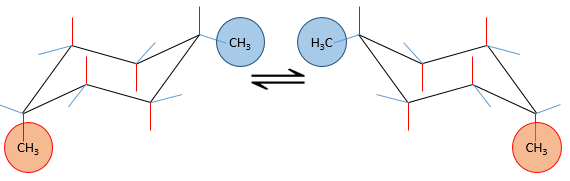
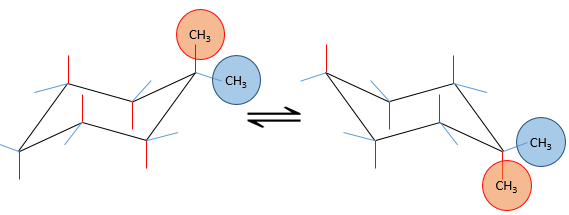
Si le second groupe est un atome de chlore à la place d’un groupe méthyle, on procède de la même manière pour savoir quel conformère est favorisé. Un groupe de chlore seule stabilise la conformation équatoriale par 2.2kJ / mol. Si les deux substituants sont équatoriaux, l’avantage énergétique totale du conformère équatorial est 7.1kJ / mol (de CH3) + 2.2kJ mol (de Cl) = 9.3kJ / mol. Cette molécule est donc plus stable que le méthylcyclohexane.
Si l’atome de chlore était axial, l’avantage énergétique total du conformère dans lequel le groupe méthyle est équatorial est 7.1kJ/ mol-2.2kJ/mol = 4.9kJ/mol. Le conformère équatorial avec un groupe méthyle est encore plus stable que l’autre conformère mais moins que le méthylcyclohexane.
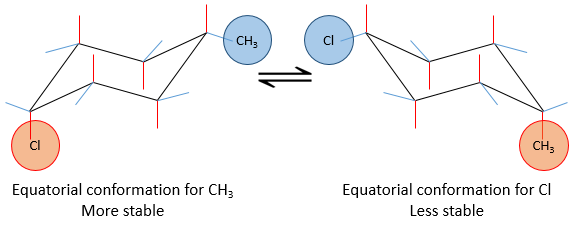
A noter que d’autres interactions peuvent affecter la stabilité des conformères. Lorsque les substituants sont sur d’autres points que 1-4, les interactions (comme l’encombrement stérique, répulsion, …) peuvent diminuer ou augmenter l’avantage énergétique d’un des conformères sur l’autre.
Alcanes polycycliques
Les molécules ne sont pas limitées à un seul cycle. Plusieurs cycles peuvent partager les carbones. Une molécule composée de deux hexanes partageant deux atomes de carbone est appelé décaline et existe dans les formes trans et cis. Les cycles ne doivent pas nécessairement avoir la même taille et un cyclohexane peuvent fusionner avec un cyclopentane.
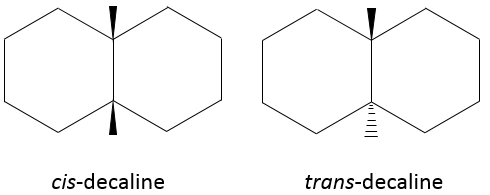
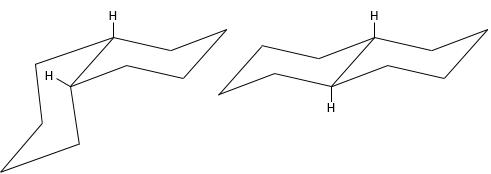
Deux cycles peuvent également être une fente dans l’autre. Cela conduit à des cycles pontés. Le Norborane est un cyclohexane ponté où 2 cyclopentanes partagent 3 atomes de carbone . Les carbones portant le pont sont appelés tête de pont.
Les alcanes polycycliques réduisent la liberté de mouvement de la molécule mais il semble qu’il n’y a pas de limite à la tension de cycle que les hydrocarbures peuvent supporter comme on peut le penser de la cyclobutane.
Les polycycliques avec des cycles de quelques atomes de carbone (3-4) ne sont en général pas produit naturellement mais peuvent être synthétisées. Tous les types de squelettes de carbone ont été réalisés au laboratoire. Ils peuvent avoir un intérêt comme explosifs s’ ils portent des groupes nitro grâce à leur tension de cycle importante .
Les polycycles de grands cycles sont souvent trouvés dans les composés naturels donnant des odeurs spécifiques, parfums, couleurs, ou des rôles très spécifiques comme hormones.
cet article a été traduit de l’anglais (cf borzuya en anglais)
Chapitre 3d : groupes fonctionnels
Autres que les alcanes, les alcènes, les alcynes et les halogénoalcanes, il existe un grand nombre de groupes fonctionnels que l’on peut trouver dans la chimie organique. En cette première année nous allons seulement jeter un coup d’oeil sur la richesse de la la chimie organique à travers les groupes fonctionnels qu’une chaîne d’hydrate de carbone peuvent porter et de certaines de leurs propriétés. Nous n’ allons pas encore parler de leur réactivité ou de la façon de les produire.
Fondamentalement les principaux atomes dans le biosphère et en chimie organique sont le C, H, O et N. Les groupes fonctionnels sont habituellement faits de O et/ou de N mais parfois aussi d’autres atomes.
Les groupes basés sur O
Il ya beaucoup de groupes fonctionnels qui contiennent des atomes d’oxygène. Les groupes contenant le N et le O seront étudiés dans le chapitre consacré aux groupes basé sur N .
L’oxygène est capteur inductif et donneur mésomérique. Capteur inductif signifie qu’il attire les électrons des atomes de carbone adjacents en raison de son électronégativité. Donneur mésomérique signifie que l’oxygène peut partager une seule paire (électrons) avec le reste de la molécule, pour former finalement des structures de résonance.
Les alcools :
les alcools sont des molécules portant un groupe OH sur un carbone.
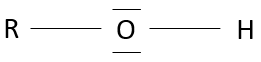
Nomenclature : ajouter -ol précédé de la position du groupe. Par exemple l’éthanol est le propan-2-ol, éthane-1,2-diol.
Les alcools peuvent être subdivisés en trois groupes en fonction du nombre de chaînes sur le carbone portant l’alcool :
Alcool primaire : Une chaîne est liée à l’atome de carbone. La formule générale est R-CH2OH.
Alcool secondaire: deux chaînes sont liées au carbone. La formule générale est R1R2-CHOH.
Alcool tertiaire : Trois chaînes sont liées au carbone. La formule générale est R1, R2, R3-COH.
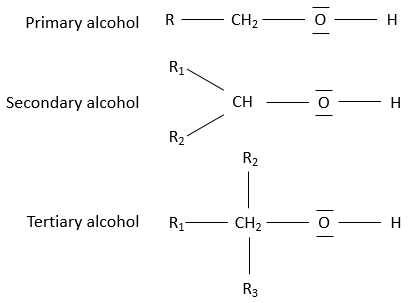
propriétés :
Les alcools sont des acides faibles (pKa ~ 16-18). L’acidité diminue des alcools primaires aux alcools tertiaires parce que les chaînes partagent leurs électrons par effet inductif vers l’O. La charge de la base conjuguée de l’alcool est donc augmentée par la présence de chaînes sur le carbone. Une plus grande charge signifie que la base conjuguée est moins stable et sera probablement moins produite par la dissociation du proton, par conséquent la diminution de l’acidité. En outre les chaînes augmentent la taille de la molécule ce qui diminue la solvatation de l’ion, cela diminue aussi l’acidité de l’alcool.
Ils sont de bons nucléophiles : les paires libres de l’oxygène peuvent attaquer des cibles électrophile.
Les alcools peuvent accepter et faire des liaisons hydrogène.
Les éthers :
un éther est un groupe dans lequel un atome d’oxygène est relié à une seconde chaîne : R1-O-R2. Remarquez R1 peut être identique à R2 ce qui donne une chaîne cyclique par un hétéroatome (= O).
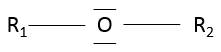
Deux nomenclatures sont utilisées :
-les noms des chaînes puis de l’éther. Exemples : éther méthylique, l’éther méthylique d’éthyle
-la forme «alcoxy alcane : La plus petite chaîne est la première noté. Exemples: méthoxyméthane, méthoxyéthane, éthoxypropane
propriétés
En général les éthers sont volatils et ont un point d’ébullition bas.
Ils peuvent être protonés, puis former un bon groupe partant pour une SN2. Les éthers sont souvent utilisés pour protéger un groupe que nous ne voulons pas remplacer: former l’éther, procéder à la substitution dans des conditions de base, et éliminer l’éther.
Les cétones :
une cétone est une double liaison entre un atome de carbone et un atome d’oxygène au milieu d’une chaîne : R1-C (= O) -R2
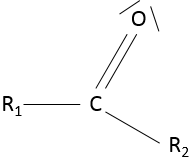
Nomenclature : le nom de l’alcane correspondant avec le suffixe one. Par exemple CH3COCH3 : propan-2-one. Notez que pour ce composé particulier un nom commun est préféré : acétone.
propriétés
Il y a un dipôle entre le C et le O de la cétone. Le C est électrophile alors que O est nucléophile. L’oxygène peut également accepter des liaisons hydrogène.
Tautomérie céto-énolique : si un hydrogène est présent sur un carbone voisin de la cétone alors une forme de résonance existe pour la cétone, acide catalysé dans des conditions basiques
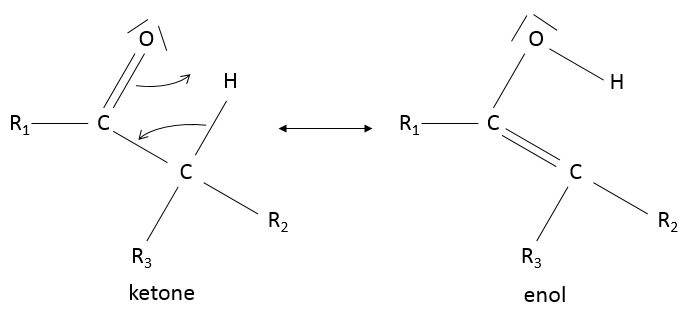
En raison de ce processus, les cétones sont acides (pKa ~ 20) : la forme de résonance stabilise la charge négative de la base conjuguée.
Aldéhyde :
un aldéhyde est similaire à une cétone sauf que c’est à la fin d’une chaîne :
RC (= O) -H.
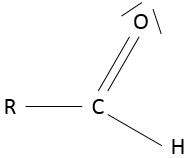
Nomenclature : pour les chaînes alcanes simples le nom de l’alcane correspondant avec le suffixe (-al) ou -carbaldehyde pour des molécules plus complexes. Si un autre groupe fonctionnel est déjà responsable du suffixe du nom (un alcool par exemple) le préfixe formyle est utilisé à la place. L’extrémité COH est également appelé un groupe formyle et le plus petit aldéhyde HCOH est appelé formaldéhyde.
Acide carboxylique :
un acide carboxylique est une cétone qui est lié à un alcool : RC (= O) -OH
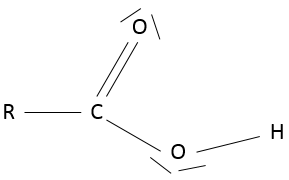
Nomenclature : le nom de l’alcane correspondant avec le suffixe (oïque) et préfixe acide. Exemples : l’acide butanoïque, l’acide propanoïque. Cependant il y a plusieurs noms communs pour certains acides carboxyliques provenant de ce que nous avons vu pour les groupes fonctionnels précédents : l’acide formique (HCOOH, de piqûres d’insectes), de l’acide acétique (CH3COOH), l’acide butyrique (CH3 (CH2) 2COOH à partir de beurre ) et beaucoup d’autres.
La base conjuguée d’un acide carboxylique est un carboxylate.
propriétés
De toute évidence les acides carboxyliques sont des acides. Ils sont plus acides que les alcools et très acides par rapport à d’autres composés organiques. Bien que les alcools ont un pKa d’environ 16 à 18 c-à-d inférieure à celui du pKa de l’eau, l’acide carboxylique est un acide faible avec un pKa compris entre 3-5. Cette acidité provient des formes de résonance du carboxylate, la base conjuguée de l’acide:
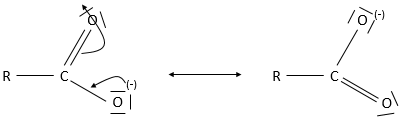
Les acides carboxyliques sont polaires et solubles dans l’eau (et les solvants protiques polaires) mais également dans les solvants non polaires : deux acides peuvent former un dimère non polaire par une liaison hydrogène.
Les esters :
un ester est la combinaison d’un acide carboxylique et d’un alcool (et c’est ainsi qu’on les produit techniquement) : R1-C (= O) -O-R2.
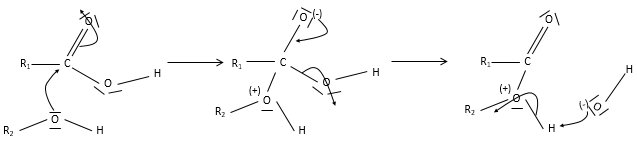
Nomenclature : la chaîne principale est celle qui porte le carboxylate
(RC (= O) -O-) et est nommé avec le suffixe -oate et le reste de la chaîne est un groupe. Exemples : Ethylethanoate (ou acétate d’éthyle), propyloctanoate.
propriétés
Les esters sont polaires et peuvent accepter des liaisons hydrogène.
Les peroxides :
il n’ est pas vraiment un type de groupe mais plutôt comme une modification des groupes existants caractérisée par la liaison de deux atomes d’oxygène.
Le peroxyde lui-même est la modification d’un éther : R1-OO-R2.
![]()
L’ hydroperoxyde est le peroxyde d’alcools : ROOH.
![]()
Le peracide (ou peroxyacide) est le peroxyde d’acides carboxyliques :
RC (= O) -OOH.
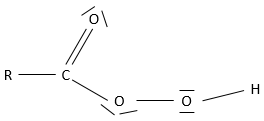
Le perester est le peroxyde d’esters : R1-C (= O) -OO-R2.
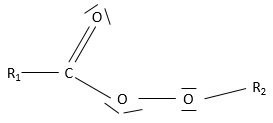
Les groupes basés sur N :
comme l’oxygène l’azote est un accepteur inductif et un donneur mésomère.
Les amines :
un groupe amine est un atome d’azote lié à une ou plusieurs chaînes carbonées. Les amines primaires sont liées à une chaîne, les amines secondaires à deux chaînes et les amines tertiaires à trois chaînes de carbone. Il existe aussi des amines quaternaires qui portent une charge positive sur l’atome d’azote . Les amines secondaires et tertiaires peuvent être trouvées dans les cycles. La pipéridine qui est très utilisée est un exemple d’amine cyclique.
Nomenclature:
on utilise le préfixe amino ou le suffixe amine. Exemple : 2-aminohexane et méthylamine.
propriétés
Ce sont de bons nucléophiles, en fonction du nombre et de la longueur des chaînes liées. Sauf l’amine quaternaire l’azote a une paire libre et peut osciller entre deux conformations.
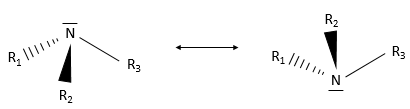
Les amines sont des bases : la paire libre peut accepter un proton. La basicité dépend de l’encombrement stérique (et donc de solvatation de l’ion) et sur la nature des chaînes. Les chaînes alkyles améliorent la basicité (par induction) tandis que les chaînes aryle (une chaîne aromatique, voir chapitre suivant) diminuent la basicité (par donation mésomère).
un groupe nitro :
un groupe nitro est composé d’un atome d’azote et de deux atomes d’oxygène. On le trouve principalement sur les cycles aromatiques par addition électrophile (à voir l’année prochaine) et non pas par substitution nucléophile.
Une charge positive et une charge négative sont présentes sur l’azote et sur l’un des deux atomes d’oxygène. En fait il existe deux formes de résonance pour le nitro et les atomes d’oxygène et elles sont équivalentes.
propriétés
le groupe nitro donne des composés explosifs. L’explosif le plus connu est TNT (trinitrotoluène). Ne pas chauffer trop ou trop vite un composé contenant des groupes nitro surtout s’ il y en a plus d’un sur le substrat.
Les amides :
les amides sont l’équivalent d’un ester sauf que l’un des oxygènes est remplacé par un azote : R1-C (= O) -NH-R2.
Nomenclature : le suffixe d’amide est simplement ajouté à la fin du nom du composé. L’équivalent d’acide acétique est l’acétamide.
propriétés
Les liaisons d’hydrogène sont possibles sur l’azote dans les deux directions. La solubilité des amides dans des solvants protiques est bonne. La solubilité est plus faible si l’azote est lié à deux chaînes de carbone (amide tertiaire).
Les amides interviennent comme l’ossature des peptides :
ils ont une structure de résonance séparant les charges.
Les nitriles :
les nitriles sont le groupe fonctionnel composé de C = N. Dans la chimie inorganique ce groupe est appelé cyanide et est très toxique.
Nomenclature : le préfixe cyano est ajouté au nom du composé.
propriétés
Ils sont utilisés pour produire des acides carboxyliques par hydrolyse, ou des amines par hydrogénation catalytique.
Ils sont aussi souvent utilisés pour étendre une chaîne carbonée.
Le carbone des nitriles est électrophile.
Les sulfonates et phosphates :
les préfixes sulfo ou phospho sont ajoutés au nom du composé
Chapitre 3 e: réactions nucléophiles
Dans ce chapitre nous allons discuter des types de réactions qui peuvent être effectuées sur les composés organiques. Dans ce cours de la première année nous nous concentrerons uniquement sur l’une des réactions les plus utilisés : la substitution nucléophile (SN).
La réaction de substitution est en effet la base de la chimie organique : son but est de remplacer un groupe d’une molécule donnée par une autre désirée. D’autres types de réactions sont l’addition et l’élimination (ils seront vus dans les cours de deuxième année).

A la fin nous pouvons, en théorie, faire ce que nous voulons avec les composés organiques. Pour l’instant vous avez déjà la moitié de l’explication du nom de la substitution nucléophile.
Un nucléophile est un composé, riche en électrons, qui est attiré par le noyau (pauvre en électrons) ainsi ils peuvent partager une paire de ces électrons pour former une liaison. Par opposition un électrophile est un composé, pauvre en électrons, qui est attiré par les électrons. D’autre part l’acidité, comme nous l’avons vu précédemment, n’est pas importante dans ces réactions. L’acidité est importante dans des solutions aqueuses mais les composés organiques ne se dissolvent pas bien dans une solution aqueuse. Dans les solutions aqueuses les espèces sont dissociées alors que dans les solutions organiques elles ne le sont pas. Les procédés de séparation tirent parti de cela pour purifier des composés distincts.
Lors d’une substitution nucléophile un nucléophile attaque une molécule cible pour substituer un groupe appelé groupe partant (LG). La cible de l’agent nucléophile est un atome de carbone pauvre en électrons car il appartient à un groupe d’atome plus électronégatif. En général les cibles sont limitées aux halogènes et aux groupes O ou N (les groupes nitro n’en font pas partie parce que l’azote a une charge positive dans ce cas). Dans le cas de SN le groupe partant prend une paire d’électrons avec lui.
Le groupe partant est un nucléophile lui-même et pour que la réaction soit effectuée il faut que le nucléophile attaquant soit plus fort que le groupe partant. Plusieurs paramètres sont déterminants dans la force nucléophile (ou nucléophilie).
Charge du nucléophile
Tout d’abord un nucléophile n’a pas à être chargé, le fait de posséder un doublet libre d’électrons est suffisant. H2O, NH3, CH3OH sont tous de bons nucléophiles grâce à leurs paires d’électrons libres (sur le O et sur le N). Cela dit, leur équivalent chargé négativement est un nucléophile plus fort.
![]()
Soit la charge ou la paire d’électrons libres doivent être disponibles pour la réaction. Si la charge (ou la paire libre) est délocalisée la nucléophilie diminue de manière significative. Malgré leurs paires libres et leurs charges les carboxylates ne sont pas de bons nucléophiles parce que leurs charges peuvent être déplacées. On dit qu’un carboxylate est stabilisé par résonance parce que ce composé a une tendance moindre à réagir avec d’autres composés en raison de ses deux formes de résonance.
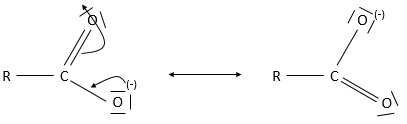
Basicité
Même si l’acidité est moins pertinente dans des solutions organiques, il existe une bonne relation entre la basicité d’un composé et sa force nucléophile.
L’électronégativité
La nucléophilie décroît avec l’électronégativité de l’atome portant la charge/ doublet. Rappelez-vous que l’électronégativité est la capacité de l’atome de garder ses électrons. Par conséquent l’atome électronégatif ne partagera pas ses électrons pour former une liaison.
Polarisabilité
nucléophilie décroît avec la polarisabilité de l’atome. Alors qu’il est contre-intuitif en ce qui concerne la réaction, ce rapport est dû à la solvatation de l’agent nucléophile. Dans les solvants protiques les espèces nucléophiles sont entourées par les molécules de solvant en formant des liaisons hydrogène avec les doublets libres de l’agent nucléophile. Fait intéressant la solvatation est plus forte pour les petites molécules que pour les plus grandes parce que la densité de charge est plus grande pour les petites molécules.
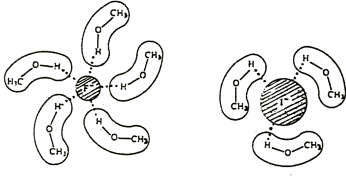
Dans les solvants aprotiques cet effet n’existe pas et la polarisabilité favorise la nucléophilie.
Mécanismes de substitutions nucléophiles :
Il existe 3 mécanismes possibles pour une substitution :
– tout d’abord l’attaque du substrat par l’agent nucléophile puis l’éjection du groupe partant
– attaque simultanée du nucléophile et le départ du groupe partant.
– tout d’abord le départ du groupe partant et ensuite l’attaque par le nucléophile.
Le premier mécanisme doit rapidement être ignoré car la première étape mène à un atome de carbone pentavalent. Les deux autres mécanismes existent et sont en concurrence. Dans certaines conditions les deux peuvent se produire en même temps. Nous allons maintenant voir les mécanismes en détail et les différences entre eux.
SN2 : Substitution nucléophile bimoléculaire
SN2 est le deuxième mécanisme de la liste : attaque simultanée du nucléophile et le départ du groupe partant. L’ensemble du processus est effectué en une seule étape et la vitesse de la réaction dépend de la concentration des deux réactifs (substrat et le nucléophile). C’est la raison pour laquelle ce mécanisme est appelé bimoléculaire.
Notre première étape est de déterminer où le nucléophile attaque sur le substrat. Imaginez la substitution d’un atome de chlore par un alcool sur une molécule donnée (X, Y et Z étant le reste de la molécule), le nucléophile (OH-) a deux options :
Attaquer par le côté du chlore ou par l’autre côté
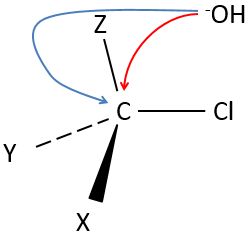
Les deux parties donnent le même résultat dans sa composition mais la connectivité diffère.
L’ attaque par le côté du groupe partant (appelé attaque frontale)
Pour être simultanée la liaison entre le substrat et le groupe partant desserre alors que la liaison entre le substrat et le nucléophile se forme. Il donne lieu à un complexe transitoire pour obtenir ensuite l’alcool substitué.

La connectivité de la molécule formée est identique à celle de la molécule de départ : X, Y et Z ne changent pas de position pendant le processus.
L’attaque par l’autre côté (appelé attaque dorsale)
Encore une fois un complexe transitoire est formé. Ce complexe montre des différences en ce qui concerne le complexe transitoire observé par une attaque frontale : la conformation de la molécule change au cours du processus. Les liaisons entre X, Y et Z avec C sont dans un plan, perpendiculaire au plan des «liaisons» avec OH et Cl ..
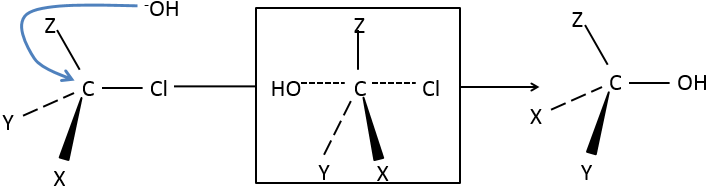
En conséquence, les positions de X et Y sont inversées. Les produits des substitutions frontale et dorsale sont des produits chiraux (images en miroir l’un de l’autre).
Nous pouvons donc facilement déterminer quels énantiomères sont obtenus en utilisant l’activité optique de chaque énantiomère. Elle a montré que seulement la substitution dorsale est produite. L’attaque frontale ne se produit donc pas.
Il ya deux conséquences :
– il ya une inversion de configuration après une SN2.
– l’encombrement stérique ralentit ou bloque la réaction. La taille de X, Y et Z joue donc un rôle important dans la vitesse de la réaction. Par exemple la SN2 est 145 fois plus rapide pour CH3Br que pour CH3CH2Br qui lui-même, est 128 fois plus rapide que pour (CH3).
SN1 : substitution nucléophile monomoléculaire
Dans ce mécanisme le départ du groupe partant intervient en premier et ensuite le nucléophile attaque le substrat.
Il y a donc deux étapes dans ce mécanisme. La vitesse globale de la réaction est donnée par l’étape la plus lente appelé l’étape limitante ou étape cinétiquement déterminante qui est le départ du groupe partant. En effet, elle implique la rupture d’une liaison et une séparation des charges. Un solvant protique aide à séparer le groupe partant et le carbocation (cation carbonique) par solvatation des espèces chargées. La deuxième étape est beaucoup plus rapide : le nucléophile est attiré par ce carbone portant la charge positive.
L’étape limitante est donc la première étape de la substitution où le groupe partant est dissocié du substrat. Cette réaction implique un seul réactif le nucléophile agit uniquement à la deuxième étape de la substitution. Le nom de SN1, substitution nucléophile monomoléculaire vient de là. La cinétique dépend alors uniquement de la concentration du substrat contrairement à la SN2 où la concentration du nucléophile a une influence sur la vitesse de réaction. Une façon de déterminer si une réaction de substitution est SN1 ou SN2 est d’effectuer une analyse de la cinétique de la réaction et de voir si la concentration du nucléophile a ou n’a pas d’influence sur la vitesse de réaction.
Pour avoir une SN1 le carbone chargé positivement, appelé carbocation, doit être stabilisé.
Carbocation
Un carbocation est un carbone portant une charge positive. Il dispose de trois liaisons dans un seul plan et une orbitale vide perpendiculaire à ce plan.
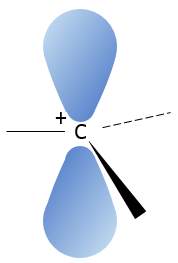
Cette configuration géométrique stabilise le carbocation avec des angles optimisés entre les liaisons (nb : une structure en forme d’avion ou quasi avion doit être obtenu) mais aussi par hyperconjugaison. Hyperconjugaison est l’interaction des électrons dans une liaison sigma (généralement C-H ou C-C) avec une orbitale p vide non liant adjacente (ou partiellement remplie) (le cas de carbocations), orbitale σ ou π antiliante ou orbitale π rempli pour donner une orbitale moléculaire étendue qui augmente la stabilité du système. En d’autres termes les liaisons des atomes de carbone adjacents aux carbocations stabilisent les carbocations lorsqu’elles sont orientées correctement.
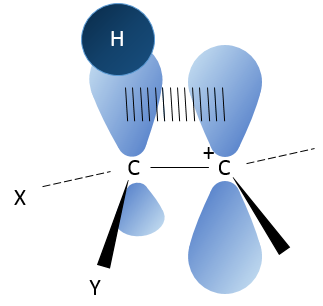
Dans l’image ci-dessus on peut voir l’hyperconjugaison d’une liaison sp entre C-H avec l’orbitale p vide du carbocation.
En conséquence les carbocations substitués sont stabilisées par hyperconjugaison. La séquence de stabilité pour carbocations est donc :
![]()
Comme vous avez peut-être déjà compris un paramètre pour déterminer si le mécanisme de substitution est un SN1 ou SN2 est la substitution du carbone portant le groupe partant. Des atomes de carbone trisubstitués conduisent à SN1 et les carbones monosubstitués ou nonsubstitués conduisent à SN2. Avec les carbones bisubstitués les deux mécanismes peuvent prendre place et sont assez lents.
Les liaisons π adjacentes les stabilisent également par (hyper) conjugaison des orbitales et par résonance.
![]()
Revenons au mécanisme de SN1. Les carbocations sont planes et peuvent être attaqués par les deux côtés du plan. Les proportions d’attaque de chaque côté sont égales sauf s’il existe une différence d’encombrement stérique pour un côté.
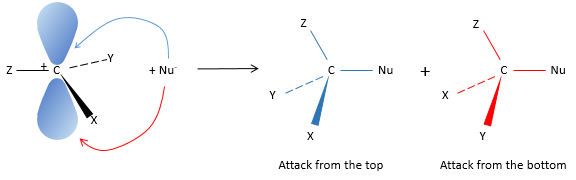
À la suite de la SN1, on obtient un mélange racémique des deux énantiomères.
Cependant nous pouvons parfois obtenir des produits très différents et inattendus d’un SN1. Par exemple :
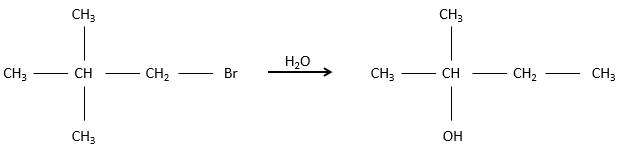
ce produit est tout à fait inattendu : le groupe partant n’a pas été substitué par un nucléophile mais plutôt par un groupe méthyle tandis que le nucléophile a pris la place d’un groupe méthyle même pas sur le même carbone que le groupe partant. La raison pour laquelle un tel produit est obtenu est que le carbocation produit au cours de la SN1, n’a pas été le carbocation le plus stable que la molécule pourrait avoir. Grâce à un mécanisme, pas totalement compris (appelé mécanisme de Wagner-Meerwein), un groupe de migrants (un méthyle ou un hydrogène) est déplacé pour obtenir une meilleure carbocation :
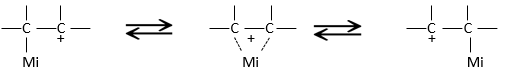
si un groupe méthyle et un hydrogène étaient sur le carbone adjacent, l’hydrogène est déplacée car elle conduit à une meilleure carbocation.

Seulement alors le nucléophile attaque le carbocation.
SN1 vs SN2
Les mécanismes eux-mêmes ne sont pas compliqués, mais il peut être difficile de déterminer quel mécanisme a lieu. Nous pouvons reprendre les paramètres influençant le mécanisme de substitution comme suit :
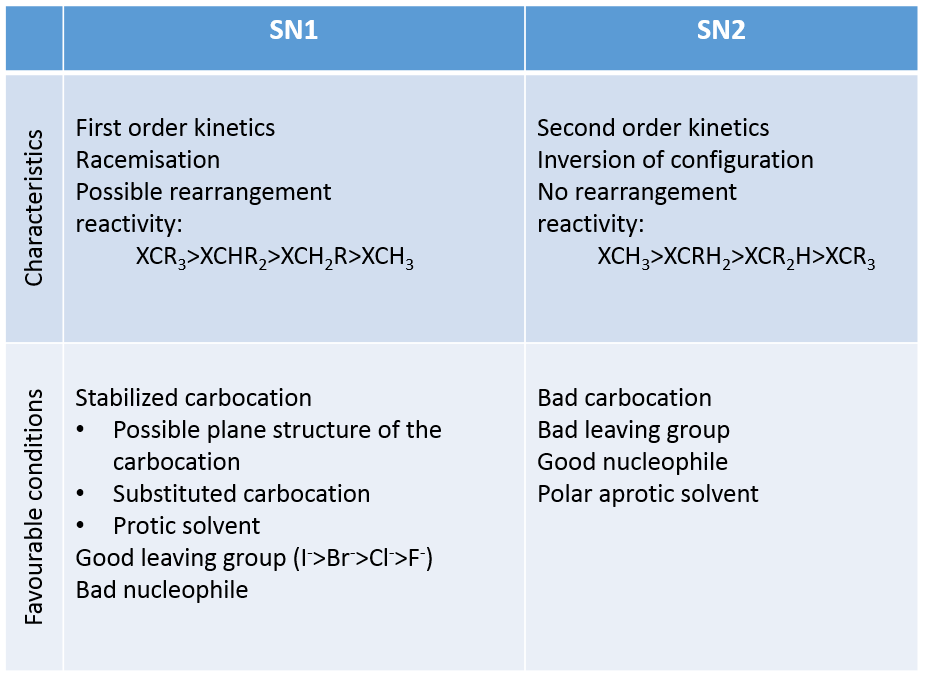
Comme il a été dit au début de ce chapitre, les substitutions nucléophiles sont l’une des réactions les plus importantes dans la chimie organique. Fondamentalement vous aurez envie de remplacer un groupe pour étendre une chaîne, de placer un groupe désiré sur le substrat ou de protéger un groupe.
D’autres substitutions que SN1 et SN2 existent mais nous les verrons ultérieurement.
Chapitre 3b: les alcènes et les alcynes
Les alcènes:
Les alcènes sont des composés organiques à base de carbone et des atomes d’hydrogène. Contrairement aux alcanes, qui ont les mêmes composants, la formule générale des alcènes n’est pas CnH2n + 2. Dans un alcène, deux atomes de carbone sont liés par une liaison pi (également appelé double liaison). Le plus petit alcène est l’éthylène CH2 = CH2.
Pour former cette liaison, chaque carbone partage deux électrons, une pour la liaison sigma et une pour la liaison pi. Il ya donc 2 hydrogènes en moins sur un alcène (que sur un alcane) dans lequel une seule liaison pi est présente. La formule générale est donc CnH2n pour un monoalcène et le nombre d’hydrogène est réduite de deux pour chaque liaison pi supplémentaire.
propène est CH2 = CH-CH3 et CH2 = C = CH2 est l’ allène.
Pour des molécules plus grandes, la position de l’alcène doit être signifiée dans la formule. L’alcène est lié à deux atomes de carbone de la chaîne et qui est toujours dans la chaîne principale, même si une chaîne plus longue peut être trouvée.
Par exemple :
est le but-1-ène. La nomenclature : le suffixe -ane des alcanes est éliminé et est remplacé par le suffixe-ène, elle-même précédée par la position de l’atome de carbone portant le groupe fonctionnel.
Un carbone partageant une double liaison est lié à trois autres atomes, et pas 4. En conséquence, la structure tétraédrique du carbone n’est plus la structure la plus stable à adopter dans le cas d’alcènes. Pour avoir une distance optimale entre les atomes, les trois liaisons sont dans le même plan. L’angle entre les liaisons est proche de 120°. La double liaison prend juste un peu plus d’espace que les liaisons simples.
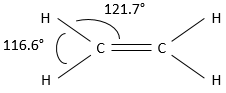
La liaison pi est moins énergique que la liaison sigma, mais elle est assez forte pour interdire la rotation d’atomes d’hydrogène autour de l’axe C = C. La liaison pi est une liaison entre les électrons sp2. Ces électrons sont situés perpendiculairement au plan de la molécule.
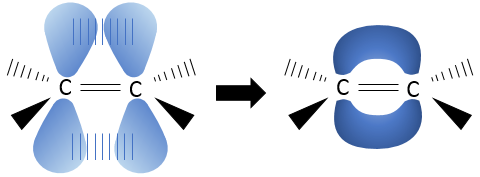
Pour former la liaison pi les orbitales électroniques sp2 doivent être alignées. Sinon les orbitales ne pourraient pas se superposer ensemble et seraient trop éloignées pour former la liaison pi, la rotation n’ est donc plus possible.
Cela signifie également que les atomes (chaînes) reliées à la double liaison restent dans une position fixe les uns par rapport aux autres autres et que nous pouvons nommer spécifiquement les composés en fonction de leur connectivité.
Prenons un exemple pour expliquer comment nommer les alcènes avec des substituants sur la double liaison.
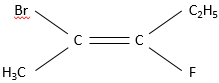
Cette alcène porte un brome, un fluor et le reste de la chaîne carbonée. Pour nommer cette substance nous regardons les atomes directement liés aux atomes de carbone portant la liaison pi. Une priorité est donnée pour chacun des deux atomes de carbone, elle va à l’atome avec le poids atomique le plus grand, dans ce cas, Br> F> C. Concernant la priorité entre les atomes de carbone, nous regardons les atomes qu’ils portent et, encore une fois, la priorité va au carbone portant l’atome de poids atomique le plus grand. La priorité est donc Br> F> C2H5> CH3.
Les substituants sont en position cis lorsque ceux-ci tout en ayant la priorité supérieure, sont situés du même côté de la liaison pi, sinon la position est trans.
Cette alcène par exemple
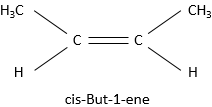
est appelée cis-but-2-ène tandis que le trans-but-2-ène est cette molécule
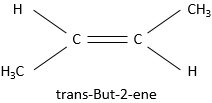
Cependant cette méthode est limitée et une méthode plus générale doit être utilisée dans le cas de molécules plus complexes comme notre exemple, nous utilisons le E, Z système. La priorité sur le carbone est laissé Br> CH3. Sur la droite de carbone, F> C2H5. Si les subsituts de priorité plus élevée sont du même côté de la double liaison la molécule a une configuration Z (pour Zusammen, ensemble en allemand). S’ils sont sur les côtés opposés de la configuration alors la configuration est dite E (pour Entgegen, en face en allemand).
La molécule de notre exemple est ainsi nommée (E) -2-bromo-3-Fluoropent-2-ène.
Exercices: nommez les molécules suivantes
cet article a été traduit de l’anglais (cf borzuya university en anglais)
Chapitre 1b: théorie atomique et réactions nucléaires
Thérie atomique
A la fin du XIX il était connu que les éléments étaient faits d’atomes, incassables et différents pour chaque élément. Les masses des atomes étaient connues pour plusieurs éléments, mais leur composition était encore un mystère.
Michaël Faraday a découvert que les atomes étaient en fait composés d’espèces chargées, même s’ils sont électriquement neutres. Sa découverte est le résultat d’une expérience dans laquelle un courant passe à travers des électrodes d’argent irrécupérables dans une solution contenant de l’argent (AgNO3). Quand le courant passe, la masse de l’électrode augmente de manière significative. Les ions d’argent dans les solutions réagissent avec les électrons du courant pour former une collection solide d’argent sur l’électrode.
![]()
Cette réaction a montré que les atomes contiennent des éléments positivement chargés ainsi que des espèces négativement chargées pour neutraliser la charge de l’atome.
Joseph John Thomson a prouvé l’existence d’électrons en 1897 au cours de ses travaux sur les tubes cathodiques. Ces tubes contiennent seulement le vide, une cathode et une anode. Si la cathode est chauffée, un courant est détecté entre les électrodes. Le chauffage détermine l’énergie cinétique transmise à l’atome de la cathode :
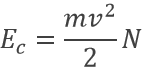
Bien que la charge e- des électrons est donnée par le courant :
![]()
L’application d’un champ magnétique H sur le tube cathodique, si seulement une petite fente permettait les électrons pour atteindre l’anode, aucun courant a été observé : le faisceau d’électrons se déviait de sa trajectoire normale selon le rapport masse / charge. Les parois de la fente ont été couvertes par ZnS une espèce fluorescente pour détecter la déviation du faisceau (rayon de déviation r) :
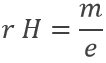
En conséquence le rapport e / m d’un électron a été déterminé :
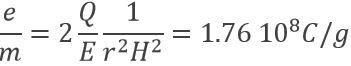
JJ Thompson a imaginé un modèle sphérique pour l’atome dans lequel une mer d’espèces chargées se déplacent (le modèle de plum-pudding).
La charge de l’électron a été déterminée par Robert Andrews Millikan. Il a envoyé un faisseau de RX sur des gouttelettes d’huile entre deux électrodes horizontales. Les gouttelettes chargées sont soumises à plusieurs forces : leur propre poids, la force électrostatique et le frottement de l’air (l’air possède une viscosité donnée).
Le poids d’une gouttelette est donnée par :
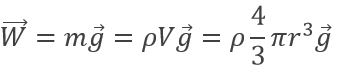
m étant la masse d’une goutte donnée qui est égale à sa densité multipliée par son volume et g est la gravité. La force électrostatique est donnée par :
![]()
où E est le champ électrique et q est la charge. Le frottement est donné par :
![]()
où η est la viscosité de l’air et v est la vitesse de la gouttelette. En l’absence de champ électrique, la goutte devrait tomber à une vitesse :
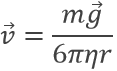
Résultant des équations de W et FR. Toutefois, dans le champ électrique E, la vitesse d’une gouttelette est affectée :
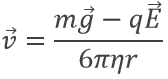
Sauf q tous les termes de la droite de l’équation sont connus et la vitesse des gouttelettes a été déterminée expérimentalement : une gouttelette est observée à travers un microscope pour mesurer le temps qu’il faut pour parcourir une distance donnée.
Le résultat de l’expérience était que la charge q était toujours un multiple de
1.602 10-19C, quelques gouttelettes étant chargées plusieurs fois. La masse d’un électron pourrait ainsi être déterminée : me=9.11 10-31kg.
Malgré son expérience brillante le modèle plum-pudding de JJ Thompson n’était pas correcte et a été réfutée par Ernest Rutherford (qui a réellement été un de ses élèves) le pionnier de la chimie nucléaire. Rutherford a étudié l’émission de particules α de l’uranium. Les particules α sont l’équivalent du noyau des atomes d’hélium : 2 neutrons et 2 protons: He2+.
Les particules α ont été envoyées vers une mince feuille d’or. Compte tenu du modèle de Thompson la totalité du rayonnement aurait dû passer à travers la feuille d’or. Il y a eu 99% de passage et il n’a pas été due à une erreur expérimentale. Certaines des particules α ont été déviées dans toutes les directions. Il a conclu que le 1% déviait sur une agrégation solide avec une charge positive intense qui composait la majorité de la masse de l’élément. Le reste du volume d’un atome est rempli par l’espace vide et un nuage d’électrons. La taille d’un atome est d’environ 1 angström (1Å)=10-10m et la taille du noyau est 10-15m.
Rutherford a également été le premier à transformer un élément en un autre. Il l’a fait en bombardant l’azote pur par des particules α et il a obtenu de l’oxygène et des noyaux d’hydrogène (les protons n’étaient pas encore connus). Il a supposé que les noyaux d’hydrogène faisaient partie du noyau solide des atomes. A partir de là les atomes n’étaient plus incassables.
Plus tard Rutherford a théorisé l’existence de neutrons pour maintenir le noyau chargé positivement en une seule pièce, en réduisant la répulsion des protons et en donnant une énergie de cohésion au noyau. Le plus gros de l’atome, les neutrons sont nécessaires (en proportion avec les protons).
Cette énergie de cohésion est en fait énorme. Par exemple le noyau de l’oxygène est formé de 8 neutrons et 8 protons mais la masse de l’atome d’oxygène est inférieure à l’addition de la masse des protons et des neutrons séparés :
![]()
La masse d’un atome d’oxygène est 2.65535.10-23g. La différence est d’environ 2.269.10-25g par atome. Comme E=mc2, nous obtenons pour une mole une énergie de cohésion de 1.23.1013J/mol d’oxygène. A titre de comparaison une réaction chimique typique est~105J/mol. Il n’est donc pas surprenant que les réactions nucléaires produisent beaucoup d’énergie.
Réactions nucléaires
La chimie nucléaire est un domaine très spécifique de la chimie. C’est un domaine où la règle de Lavoisier ne marche pas : les éléments ne sont pas conservés et la masse peut être convertie en énergie. Pourtant plusieurs types de réactions peuvent être triés. Mais d’abord nous allons jeter un oeil à la façon d’écrire les isotopes. L’isotope 238 de l’uranium est écrit :
![]()
U est le symbole de l’élément, la masse est écrite en haut à gauche de l’élément et son numéro atomique est écrit en bas à gauche.
1) La production des particules α
Rappelez-vous qu’un
![]()
La désintégration de l’uranium 238 est un exemple de réaction produisant des particules α :
![]()
La valeur totale du nombre supérieur est conservée au cours de la réaction. La même chose est vraie pour le nombre inférieur. Le produit γ est un rayonnement gamma, produit par désintégrations radioactives, car le noyau formé est généralement dans un état excité et quand il revient à son état de base l’énergie est libérée sous la forme d’un rayonnement électromagnétique.
2) La production de particules β
Les particules β sont des petites espèces chargées émises lors de certaines réactions nucléaires. Le thorium 234 obtenu précédemment peut produire ce genre de particules :
![]()
La masse de l’élément n’a pas changé au cours de cette réaction. Cependant l’élément a été modifié de thorium à Protactinium, un neutron est devenu un proton. La particule β produite au cours de cette réaction est un électron rejeté du neutron pour devenir un proton.
Un antineutrino est également généré. Un noyau a donné une rotation en fonction de sa charge : elle tourne sur elle-même dans une direction donnée. Les électrons ont également un spin.
Au cours de la réaction nucléaire en haut, la charge du noyau a changé et un antineutrino est libéré pour obtenir la bonne rotation.
Une seconde particule β peut être obtenue, par exemple, lors de la désintégration de sodium 22 :
![]()
La particule β est un électron mais pas un positron. Un neutrino est également obtenu lors de cette réaction. Les neutrinos et antineutrinos sont des rayonnements qui peuvent passer à travers tout.
Les électrons et les positrons peuvent se neutraliser pour produire un rayonnement gamma :
![]()
Fission :
La fission est faite en bombardant un isotope avec des neutrons. Dans les centrales nucléaires, la fission se fait sur l’uranium 235 :
![]()
Il ya plus de neutrons produits par la réaction que nécessaire pour le lancer. Cette réaction peut donc recommencer tant qu’il est de l’uranium 235 dans le réacteur. Un autre moyen d’arrêter la réaction est de piéger les neutrons avec un autre isotope.
Fusion
La fusion se fait par la fusion de deux isotopes. Par exemple deux isotopes d’ hydrogène peuvent produire de l’hélium :
![]()
Une autre façon d’obtenir l’hélium est de bombarder l’hydrogène avec des électrons :
![]()
Il est également une réaction de fusion.
Les éléments utilisés dans la fusion et dans la fission sont différents. L’énergie de la cohésion est différente pour chaque élément et on peut observer un maximum d’énergie de la cohésion/nucléon (proton + neutrons) pour 56Fe.
La fusion est effectuée sur des éléments de masse inférieure à 56Fe. Les atomes gagnent de l’énergie de la cohésion lors de la fusion. D’un autre côté des éléments de masse élevée perdent de l’énergie de cohésion et de la masse à partir de 56Fe.
Temps de demi-vie
Les éléments radioactifs ne restent pas toujours actif. La radioactivité diminue avec le temps proportionnellement au nombre des particules :
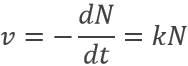
où v est la vitesse de décomposition, N le nombre de particules radioactives et k une constante de vitesse en fonction de l’isotope.
Comme elle dépend du nombre de particules la vitesse diminue au fil du temps. Nous pouvons intégrer l’équation de la vitesse :
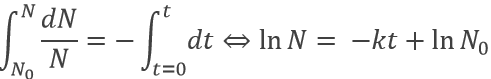
Le temps de demi-vie est le temps nécessaire pour diminuer la population d’un élément radioactif de moitié :
 ce temps ne dépendant que de l’isotope et ne dépend pas de la population de l’isotope.
ce temps ne dépendant que de l’isotope et ne dépend pas de la population de l’isotope.