La structure primaire d’une protéine est la succession, ou la séquence, de l’AA. Les protéines sont constituées d’une chaîne linéaire d’acides aminés. Cette chaîne linéaire prend une structure 3D (structures secondaires et tertiaires) en raison des interactions entre les séquences de la chaîne et les interactions (hydrophobes ou hydrophiles) avec l’environnement. L’ordre des AA et leurs quantités sont bien définis pour chaque protéine. La première structure jamais déterminée était celle de l’insuline. Cette protéine a une longueur de 51 AA, ce qui est petit en comparaison avec d’autres protéines pouvant atteindre 8000 AA de long.
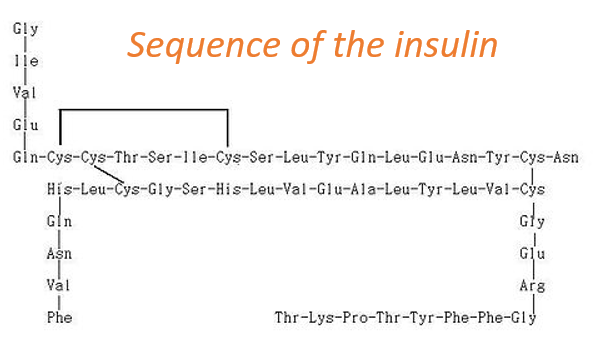
A l’exception des ponts disulfures entre les cystéines, cette protéine peut être vue comme deux chaînes linéaires avec respectivement 21 et 30 AA. Comme l’insuline n’est pas composée d’une seule chaîne, cette protéine n’est évidemment pas le produit primaire d’un ribosome. Quelques opérations supplémentaires ont été appliquées aux formes précurseurs de l’insuline pour obtenir la protéine fonctionnelle. Nous verrons ce processus plus en détail plus tard.
Pour déterminer la structure primaire d’une protéine, nous avons besoin qu’elle soit pure. Pour séparer les protéines des autres molécules, nous effectuons une dialyse: la solution contenant les protéines dans un sac avec une membrane poreuse à travers laquelle les petits ions peuvent passer mais pas les macromolécules. Une fois l’équilibre atteint, on répète le processus pour obtenir la meilleure pureté possible. Nous devons encore séparer les macromolécules et cela se fait:
par chromatographie :
– ionique: les protéines ont une charge qui dépend du R des acides aminés. La charge dépend du solvant utilisé. À un pH donné, la protéine cible est chargée positivement. Le placement d’un polymère chargé négativement dans la colonne retient la protéine pendant que les autres passent à travers la colonne. Après un certain temps, nous changeons de solvant pour délier la protéine du polymère.
– d’exclusion: les protéines sont séparées en fonction de leur taille. Les grandes protéines se déplacent plus vite que les petites car elles ne peuvent pas pénétrer dans les trous des billes composant la phase immobile.
– ligands: la protéine peut se lier avec des ligands spécifiques. Normalement, seule cette protéine devrait se lier, ce qui assure une grande pureté.
par électrophorèse:
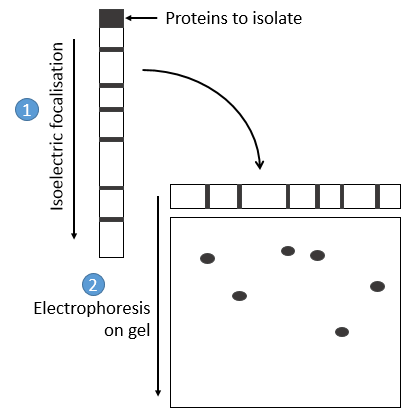
les protéines sont séparées sur la base de leur point isoélectrique: les protéines sont placées sur un gel de polyacrylamine dont le pH n’est pas uniforme mais a un gradient. Comme les protéines sont chargées en fonction du pH du solvant, il se déplace jusqu’à ce que sa charge devienne neutre. Après cela, nous prenons le gel et l’appliquons sur un second gel long et poreux où les protéines sont déplacées par l’application d’une charge et leur vitesse dépend également de la taille de la protéine. De cette façon, nous obtenons une séparation 2D des espèces.
Une fois les protéines isolées, nous pouvons commencer à déterminer sa structure. Pour ce faire, nous utilisons un exopeptidase: son rôle est de ne cliver que la liaison avant le dernier AA. Le problème est que les enzymes ne sont pas synchroniques: elles ne commencent pas toutes simultanément et certaines chaînes peuvent être clivées plusieurs fois tandis que d’autres sont clivées une fois ou pas du tout. Il peut donc y avoir un mélange d’acides aminés dans la solution et la détermination de la bonne séquence peut être difficile. Pour minimiser ce problème, l’expérience doit être très courte.
Une autre méthode (d’Edman) consiste à affaiblir la dernière liaison peptidique et ensuite à utiliser un réactif faible qui n’hydrolyse que cette liaison. Dans le cas présenté, un isothiocyanate de phényle réagit avec le résidu amino-terminal pour former un thioamide. La liaison peptidique suivante est affaiblie et peut être clivée sans hydrolyse en raison de la formation d’un cycle.
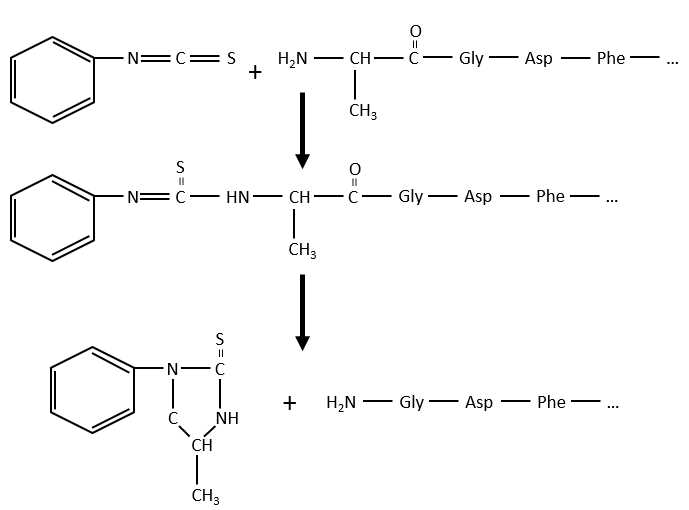
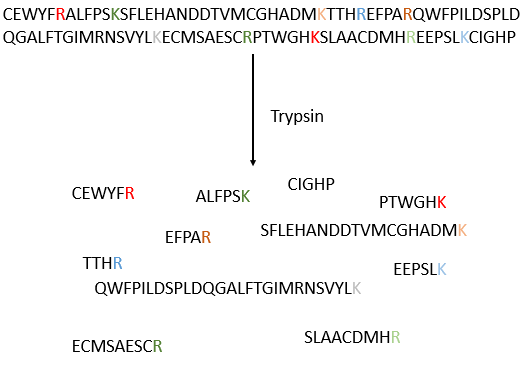
Le procédé peut être répété plusieurs fois pour déterminer la séquence amino-terminale initiale mais le rendement de la réaction n’est pas de 100%. En conséquence, il ne peut être répété que 30 fois. De plus, parfois les chaînes se rompent et nous avons donc une nouvelle extrémité qui donne des résultats différents. De toute façon 30 AA n’est pas assez bon pour déterminer la structure des protéines qui peuvent être longues de 8000 AA. L’ordre des 30 premiers acides aminés ne donne aucune indication sur la nature des acides aminés suivants. La seule façon de déterminer la structure est de commencer par fragmenter la protéine en petits morceaux. Nous allons essayer de couper la protéine seulement entre certains acides aminés pour avoir des points de référence. Il existe certaines enzymes endopeptidases spécifiques qui clivent les protéines seulement après un acide aminé particulier. Il détecte la présence d’un R spécifique, s’y lie et clive la liaison peptidique. La trypsine est une enzyme qui clive la liaison peptidique après une lysine ou de l’arginine.
Nous séparons les fragments par chromatographie et déterminons la séquence de chaque fragment (avec la méthode d’Edman par exemple). Pourtant, nous ne savons pas dans quel ordre les fragments sont dans la protéine. Le processus est répété avec une autre endopeptidase spécifique. Après plusieurs fragmentations et séquencements, nous pouvons essayer de comprendre quelles sont les séquences communes aux différentes fragmentations et reconstruire toute la séquence.
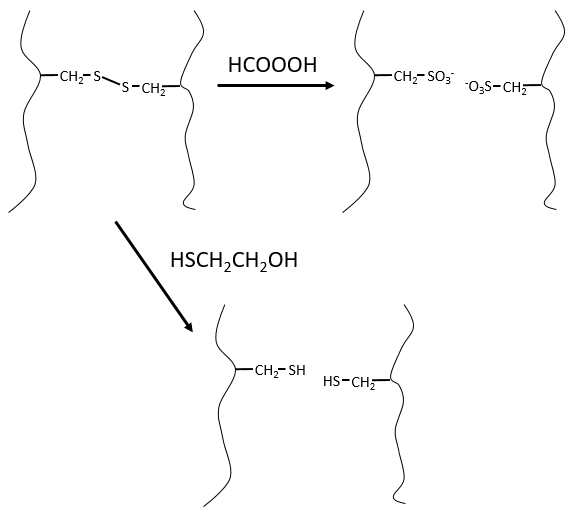
Ce serait parfait s’il n’y avait pas de liaisons SS le long de la chaîne peptidique. Il y a donc une étape qui doit être faite avant la fragmentation: nous utilisons l’acide performique (HCOOOH) pour oxyder les sulfures. Cette méthode est irréversible. Une seconde méthode, réversible, peut être utilisée à la place: le mercaptoéthanol réduit les sulfures.
Nous pouvons reprendre la structure primaire en 4 points:
la chaîne est linéaire et non ramifiée.
entre les acides aminés, les seules liaisons covalentes sont les liaisons peptidiques et les liaisons SS entre les cystéines. Les autres interactions sont l’attraction / la répulsion entre les groupes chargés et les liaisons H.
la séquence est complètement définie et arbitraire: une partie de la séquence ne définit pas le reste de la séquence. Par exemple, si on connaît 10 acides aminés, on ne peut pas dire que les 5 suivants sont une guanine, une cystéine, etc …
les protéines de différentes espèces sont similaires mais pas identiques. Il y a quelques fragments clés qui sont communs mais la structure entière peut varier. Une exception est l’histone. Cette protéine est commune à toutes les espèces eucaryotes: c’est la protéine responsable de la compaction de l’ADN dans le noyau cellulaire. Même si l’information contenue dans l’ADN diffère d’une espèce à l’autre, sa structure est identique et la protéine responsable de son enroulement/déroulement est donc la même pour toutes les espèces eucaryotes.
Certaines protéines fixent un (ou plusieurs) monosaccharide. Ce processus est appelé glycosylation et conduire à une glycoprotéine.
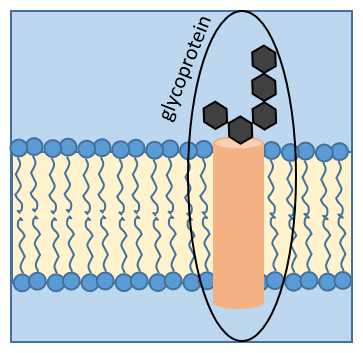
Le monosaccharide peut être fixé sur un atome d’azote (glycosylation N-liée, exclusivement sur l’asparagine) ou sur un atome d’oxygène (glycosylation O-liée). Le monosaccharide peut lui-même être lié à une chaîne de monosaccharides. Nous observons fréquemment des glycoprotéines dans les membranes où elles jouent le rôle de récepteurs. La présence du monosaccharide sur la protéine permet une grande diversité dans les récepteurs pour les cellules.