L’état des composés dépend de la température et de la pression. Nous cartographions les états de la matière sur le diagramme de phases. La pression est mise en ordonnée et la température en abscisse :
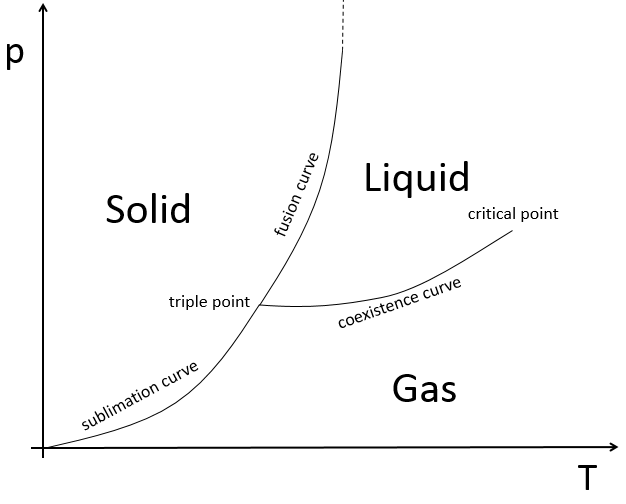
Les états sont séparés par des traits pleins ce sont les courbes de transition de phase. La courbe entre le liquide et le solide est appelée la courbe de fusion.
La courbe entre le liquide et le gaz est appelé la courbe de vaporisation/ coexistence. La courbe entre le solide et le gaz est appelée la courbe de sublimation.
Courbe de vaporisation :
![]()
Dans un système fermé contenant un liquide et un gaz il existe un équilibre entre les deux phases. Le gaz devient liquide à une vitesse donnée vGL et le liquide devient gaz à une vitesse donnée vLG. L’équilibre est atteint lorsque les vitesses sont égales.
Le liquide devient un gaz si les molécules ont une énergie cinétique suffisamment grande. Si cette énergie n’est pas assez grande les molécules restent liquides en raison des interactions entre les molécules. L’énergie cinétique peut être fournie par chauffage ou excitation. Typiquement nous devons chauffer l’eau pour la faire bouillir.
La réaction inverse, c.à.d la transformation d’un gaz à un liquide, est favorable mais pas complète en raison de l’entropie. L’entropie est une expression du désodre dans le système. La deuxième loi de la thermodynamique impose que l’univers évolue vers un plus grand désordre. Cela signifie que la somme des variations d’entropie à l’intérieur et à l’extérieur du système doit être positive :
![]()
Il existe donc une compétition entre l’augmentation de l’entropie et la diminution de l’énergie dans le système.
L’enthalpie libre de Gibbs représente/reflète cette compétition :
![]()
L’enthalpie libre de Gibbs est donc un potentiel thermodynamique tenant compte des implications du système sur son environnement.
La pression de vapeur dans le système à l’équilibre p° (également appelée la pression de Clausius-Clapeyron) est :
![]()
Les termes entropiques sont présents dans le pré-exponentiel constant et l’enthalpie est dans le terme exponentiel. Lorsque la température augmente la pression de vapeur augmente de façon exponentielle. Expérimentalement nous pouvons trouver l’enthalpie de vaporisation en traçant la courbe
ln p° vs 1/T :
![]()
La pente de la courbe est -AH / R. La valeur de ΔHvap dépend des interactions entre les molécules. Des interactions fortes font que les molécules ne sont pas facilement séparables les unes des autres et que la vaporisation nécessite plus d’énergie, soit ΔHvap est grand et p° est faible. Il est logique : si les molécules ne se vaporisent pas facilement il n’y a pas beaucoup de molécules dans le gaz et la pression de la vapeur est donc faible.
Dans le cyclohexane il n’existe pratiquement aucune interaction entre les molécules et son ΔHvap est faible. Le cyclohexane est un liquide volatil.
La température d’ébullition d’un liquide est la température à laquelle la pression de vapeur d’un liquide est égale à la pression atmosphérique. Pour l’eau cette température est de 373K (100° C) si la pression atmosphérique est de 1 atm. En altitude la pression atmosphérique diminue et il en va de même pour la température d’ébullition. Par exemple la température d’ébullition est de 363K (90° C) à une altitude de 3000 m. Dans une cocotte-minute, la pression est supérieure à 1 atm et l’eau bout à une température supérieure à 100° C.
Courbe de sublimation :
la sublimation est la transition de phase entre un solide et un gaz :
![]()
Nous pouvons définir une pression de vapeur pour les solides aussi mais il est beaucoup plus faible que pour un liquide parce que les interactions dans un solide sont plus fortes que dans un liquide. L’équation est similaire :
![]()
mais la constante est différente et ΔHsub>>ΔHvap, pour exactement les mêmes raisons.
La courbe de fusion :
la fusion est la transition de l’état d’un solide à un liquide.
![]()
On peut aussi définir une pression telle que
![]()
L’enthalpie de fusion est très faible et la courbe est presqu’une ligne droite verticale. En règle générale la pente est positive, ce qui signifie que, quand un liquide devient solide (par diminution de sa température par exemple), son volume diminue. Il est cependant pas toujours vrai. Un cas particulier est l’eau: le volume de 1 kg d’eau est inférieur au volume de 1 kg de glace. C’ est une astuce des barmens pour diminuer le volume de la boisson qu’ils donnent à leurs clients :
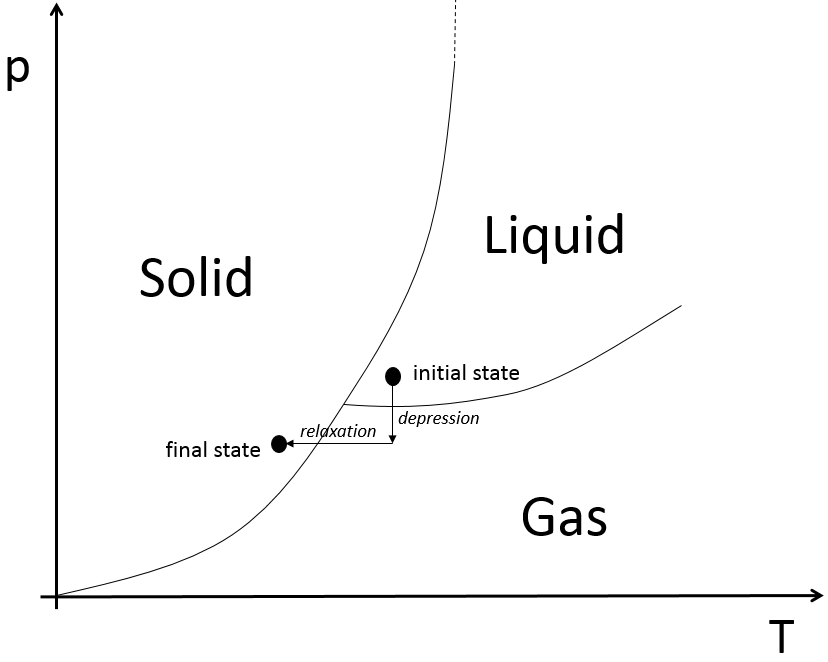
Le dioxyde de carbone dans un extincteur est à l’état liquide à proximité du point triple. Lorsque nous utilisons l’extincteur la pression chute brusquement. Nous devrions donc nous attendre à un gaz à l’extérieur du tube; Cependant il ya une détente du gaz pendant le processus (passage d’un espace étroit à un grand espace) qui implique une diminution de la température ce qui fait que le gaz devient un solide.
Variance :
c’est le nombre de degrés de liberté qui caractérisent un état :
![]()
Où C est le nombre de constituants de la matière et φ est le nombre de phases. Jetons un oeil à différents points sur le diagramme de phase :
-au milieu d’une phase : V = 1-1 + 2 = 2 : nous pouvons modifier la pression ou la température sans transition de l’Etat.
-sur une courbe : V = 1-2 + 2 = 1 : si nous voulons modifier la pression ou la température de la matière sans transition de l’Etat l’autre paramètre (T ou p) doit être fixe.
-sur le point triple : V = 1-3 + 2 = 0 : si nous voulons garder la question dans cet état, nous ne pouvons pas changer les conditions de température et de pression.
Au point triple l’enthalpie de sublimation est égale à la somme des enthalpies de vaporisation et de fusion. Le point triple de l’eau est à t = 273,16K et p = 0.006atm.
Après ce point les phases liquide et gazeuse deviennent indiscernables. Dans l’eau le point critique se produit à environ 647 K et 218 atm.
Au voisinage du point critique les propriétés physiques du liquide et de la vapeur changent de façon spectaculaire les deux phases deviennent de plus en plus similaires. Par exemple de l’eau liquide, dans des conditions normales, est presque incompressible, a un faible coefficient de dilatation thermique, a une constante diélectrique élevée et est un excellent solvant pour les électrolytes. Près du point critique l’eau devient compressible, extensible, un pauvre diélectrique, un mauvais solvant pour les électrolytes et préfère se mélanger avec les gaz non polaires et des molécules organiques.
Au point critique une seule phase existe. La chaleur de vaporisation est de zéro.
Au-dessus du point critique (T> Tc et p> pc) il y a un état de la matière qui est continuellement connecté, sans transition de la phase, à la fois avec le liquide et l’état gazeux : fluide supercritique. Un tel fluide a généralement des propriétés entre celles d’un gaz et d’un liquide.
Cas de mélanges :
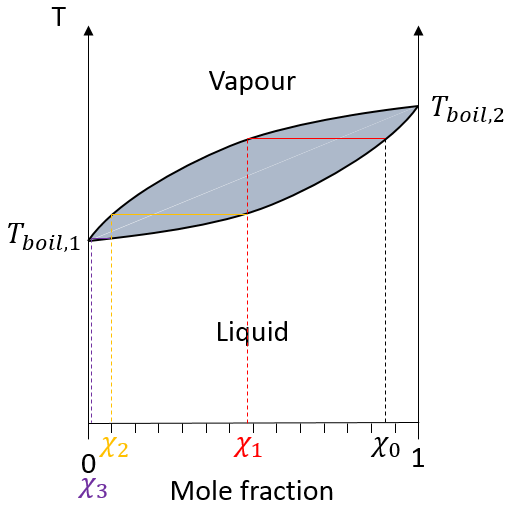
dans une solution les éléments du mélange ne doivent pas nécessairement avoir le même point d’ébullition. L’élément ayant le point d’ébullition inférieur va s’évaporer plus rapidement que les autres composants du mélange. Si l’on considère un mélange binaire, on peut tracer la composition du mélange en fonction de la température sur un diagramme montré ci-dessous :
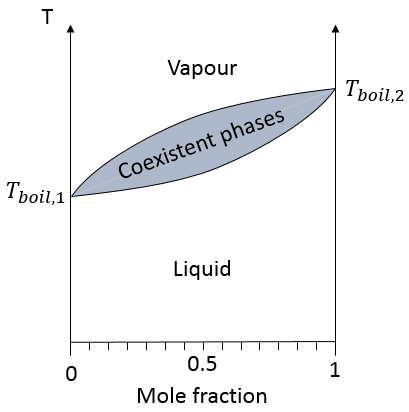
Sur ce diagramme la température est rapporté sur l’axe des ordonnées et la fraction molaire χ du deuxième composant du mélange est définie comme abscisse. A χ = 0, la solution est une solution pure du soluté 1 et la température d’ébullition de la solution est Tboil,1. A χ = 1, la solution est une solution pure du soluté 2 et la température d’ébullition de la solution est Tboil,2. Entre les deux la solution est un mélange des deux composants. Dans ce cas nous avons considéré une solution dans laquelle le soluté a une température d’ébullition plus basse que le soluté 2. Si l’on augmente la proportion du soluté 2 la température d’ébullition du mélange augmente. Cette augmentation n’est pas linéaire et on peut voir qu’il y a un domaine dans lequel, à la fois, la vapeur et le liquide coexistent. Si l’on regarde à une température donnée T la composition du liquide et de la vapeur est différente : la composition de la vapeur est constituée par une fraction plus importante du soluté qui est plus volatil que le liquide :
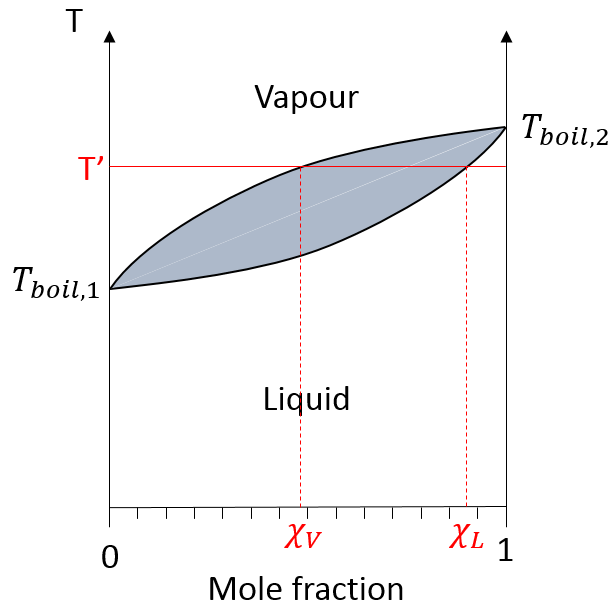
Distillation :
la distillation est un procédé de séparation basé sur la différence de température des différents solutés dans une solution bouillante. Fondamentalement si la solution initiale a une fraction molaire χL, nous pouvons faire bouillir à la température T ‘. L’objectif est de recueillir la vapeur et de la liquéfier. La solution recueillie a une composition χV. Nous pouvons continuer à faire cela plusieurs fois pour obtenir une solution quasiment pure du soluté 1 :
En pratique plusieurs distillations sont effectuées à la fois. L’installation se compose d’un ballon contenant la solution initiale un système de chauffage (comme une lampe infra-rouge), une colonne de distillation, d’un tube réfrigérant et d’un ballon pour recueillir le distillat. Les thermomètres peuvent être placés dans le premier ballon et au sommet de la colonne de distillation pour contrôler la température de la solution et de la vapeur :
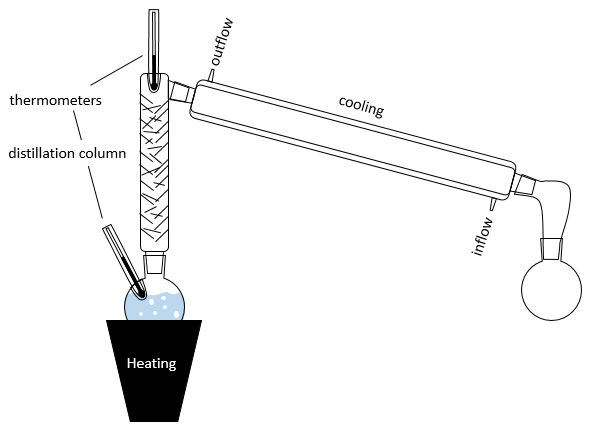
Le dispositif de refroidissement est composé de deux tubes un mince placé à l’intérieur de l’autre. Le tube interne présente des ouvertures aux deux extrémités et est relié à la colonne et le ballon de collecte. L’autre tube a des ouvertures latérales connectées à un contre-courant d’eau fraîche. Le flux d’eau fraîche va de cette façon pour éviter une zone non chargée et aussi parce que l’eau près de la colonne, qui a été chauffée par la vapeur, est rapidement évacuée. Si l’eau allait dans le sens inverse l’eau serait chauffée par la vapeur et la puissance de refroidissement du tube serait diminuée.
La colonne de distillation peut être remplie de petits morceaux/tubes de verre pour augmenter la surface de contact avec de la vapeur. Dans certaines colonnes les aiguilles font partie de la colonne et il n’est pas nécessaire d’ajouter des pièces supplémentaires.
Dans la colonne de distillation de la vapeur chaude monte et remplit les morceaux de verre de plus basse température. A leur contact il y a un échange de chaleur et la vapeur peut redevenir liquide, pas avec la composition du liquide dans le ballon χ0 (pour maintenir la notation de la figure ci-dessus), mais la composition de la vapeur χ1. Les gouttelettes finiront par retomber dans le ballon ou s’évaporer sur leur chemin à cause de la vapeur chaude. Leur composition sera maintenant χ2 comme si deux distillations avaient été effectuées. La vapeur poursuit son chemin vers le sommet de la colonne, de plus en plus pur, et entre dans le tube de refroidissement où il se transforme en liquide terminant sa course à l’intérieur du ballon de collecte.
Il est bon de mettre quelques pierres ponces dans le ballon pour éviter la surchauffe et d’obtenir des bulles d’une taille controlée.
Azéotropes :
Le schéma de l’ébullition n’est pas toujours aussi simple. Certains mélanges sont dits azéotrope. Un mélange de type azéotrope se comporte comme une solution à un composant, à savoir, la fraction molaire ne change pas au cours de l’ébullition. Sur le diagramme les courbes ressemblent à :
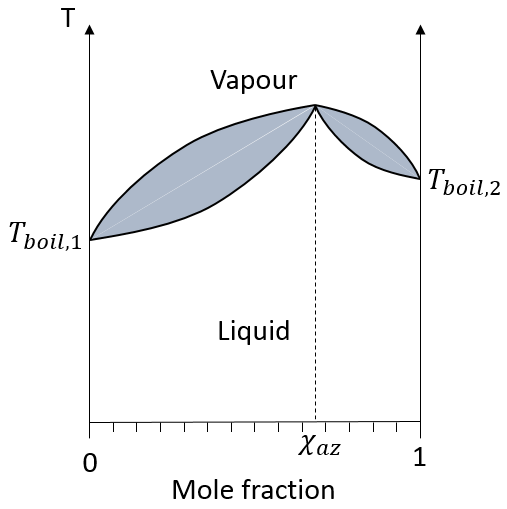
Si la solution initiale a la composition azéotrope les compositions liquide et vapeur sont identiques et ne changent pas au fil du temps. Si la solution initiale possède une fraction molaire plus faible nous allons finalement obtenir une solution pure du soluté 1 mais il est impossible d’obtenir une solution pure du soluté 2 : la composition tend vers l’azéotrope. La même chose est vraie si la composition initiale du mélange possède une fraction molaire supérieure à χaz : il sera impossible d’obtenir une solution pure du soluté 1 dans ce cas.
Deux types d’ azéotropes peuvent être triés : les aspects positifs et les aspects négatifs. Les différences sont tout simplement leur température d’ébullition. Sur le schéma ci-dessus un azéotrope positif est représenté : sa température d’ébullition est supérieure à celles des composants purs du mélange. Un azéotrope négatif a une température d’ébullition plus basse.
Démixtion :
dans les mélanges les interactions différentes existent : les interactions entre deux molécules identiques et entre les différentes molécules du mélange si
![]()
puis le mélange a tendance à se séparer. L’expression ci-dessus signifie que les interactions entre les molécules identiques sont plus favorables que les interactions entre des molécules différentes. Imaginez que, dans ce cas, les molécules A et B sont parfaitement mélangés. Les interactions entre A et B ne sont pas nécessairement défavorable mais avec le mouvement brownien, les molécules ont tendance à se regrouper entre les espèces et elles forment des grappes/bulles d’avoir le maximum d’interactions favorables. Voilà comment les émulsions se forment. Les grappes vont fusionner ensemble au fil du temps pour améliorer le rapport volume/surface des grappes et la solution finira par finir comme deux phases séparées. C’est le phénomène de démixtion :
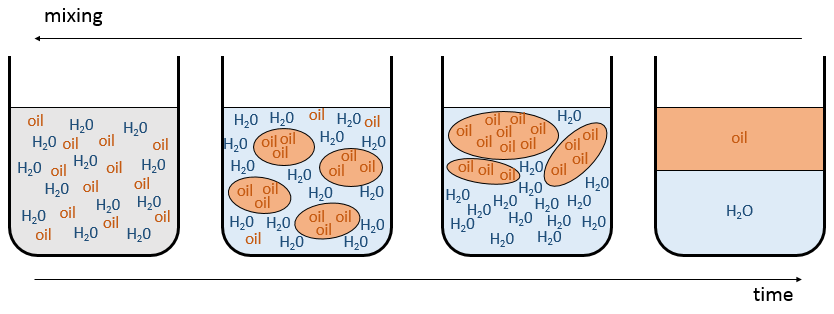
Par exemple l’huile et l’eau ne se mélangent pas bien : ils forment deux phases distinctes. Pourtant nous pouvons diluer un peu d’eau dans l’huile ou vice versa mais si nous continuons à ajouter de l’eau les phases vont se séparer comme indiqué sur la figure suivante :
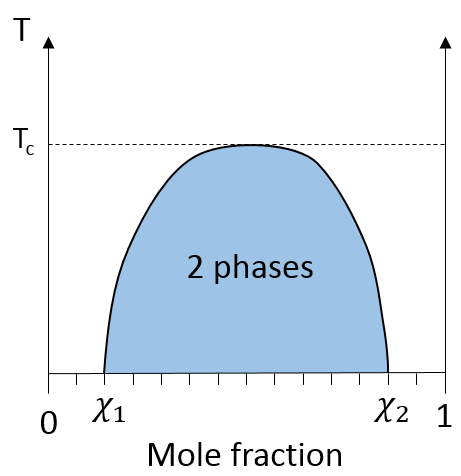
Si l’huile est la composante 1 et l’eau la composante 2, ce chiffre montre que nous pouvons ajouter de l’eau à l’huile jusqu’à la fraction molaire χ1 avant une séparation de phases. Nous pouvons ajouter de l’huile dans l’eau sans démixtion pour les fractions molaires de plus de χ2. Comme nous pouvons le voir le processus de démixtion dépend de la température. La quantité de soluté que l’on peut dissoudre avant la démixtion dépend de la température ambiante. Au-dessus d’une température critique (pas la même que dans le diagramme de phase), le mélange reste comme une phase quelle que soit la composition.
les propriétés colligatives :
Les propriétés colligatives sont des propriétés qui dépendent du nombre de particules dans le solvant, du solvant mais pas de l’espèce de soluté. Le fait que la température d’ébullition de l’eau augmente quand on ajoute un peu de sel est à l’intérieur d’une propriété colligative. Il n’a pas d’importance qu’il soit le sel ou autre chose, la température de congélation diminue aussi : nous utilisons du sel sur la neige pour la faire fondre. Dans le diagramme de phase la courbe de coexistence a été traduite vers le bas en raison de l’addition du sel. La loi ébullioscopique dit que :
![]()
Avec kb la constante ébullioscopique et ms la molalité du soluté. Lorsque nous ajoutons du sel dans l’eau nous ajoutons deux espèces : les ions Cl– et Na+. Il est donc préférable d’écrire cette relation comme suit :
![]()
ν est appelé le facteur van’t Hoff et est le nombre d’ions trouvés par la formule unitaire. En fait nous observons que ce facteur est souvent plus petit que sa valeur attendue : quelques-unes des molécules ne se dissolvent pas dans la solutio :
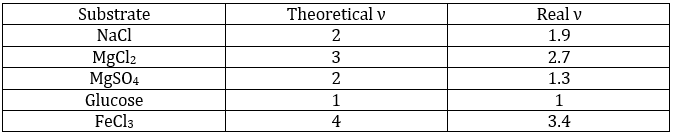
Cela signifie que pour 100 molécules de NaCl en solution, 90 se dissolvent pour produire le Cl- ions et Na+ et 10 molécules ne se dissolvent pas (soit un total de 190 molécules au lieu de 200). ν peut être déterminé à partir de la constante de dissociation de la molécule. Par exemple toutes les molécules d’un acide faible ne se dissocient pas dans l’eau.
![]()
La Ka de cette réaction est :
![]()
Jetons un coup d’oeil à la concentration des espèces avant et après la réaction. En fait, au lieu des concentrations, nous allons considérer la concentration/ concentration initiale de CH3COOH :

Le nombre total de molécules est
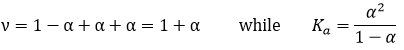
Donc nous savons qu’il ya une diminution de la température en raison des propriétés colligatives. La variation de température est couplée à une variation de la pression de vapeur du mélange. Intuitivement la pression de vapeur du sel dissout est en général très faible en ce qui concerne la pression de vapeur de l’eau. Certains sels ne sont pas volatiles du tout. La pression est égale à la somme des pressions partielles
![]()
L’addition d’un sel B dans la so