Les cycles aromatiques, tels que le benzène, sont très stables en raison de leur énergie de résonance. Par conséquent, il est très difficile d’ « ouvrir » le cycle habituel par une réaction d’addition.
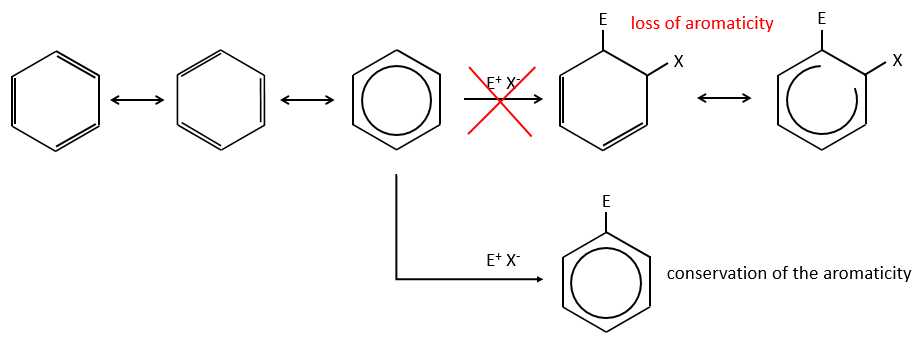
Au lieu de réactions d’addition, on observe des réactions de substitution. Le mécanisme comporte deux étapes.
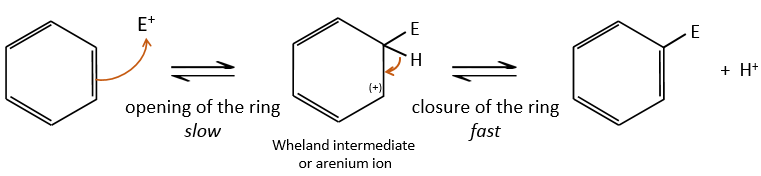
1) la première étape est l’attaque électrophile d’une liaison π sur un électrophile. Comme cette étape implique la perte d’aromaticité du substrat, cette étape est lente et l’étape de détermination de la réaction. Le produit de cette étape est appelé un intermédiaire de Wheland ou un ion arénium.
2) la deuxième étape comprend l’enlèvement d’un proton du cycle pour régénérer l’aromaticité. Cette étape est rapide.
La plupart des électrophiles doivent être activés pour permettre à la première étape de la réaction de se faire. Elle se fait par un acide de Lewis
Halogénation :
FeBr3 et AlCl3 peuvent être utilisés comme acides de Lewis pour lier un atome d’halogène (Br et Cl respectivement) sur un noyau benzénique.
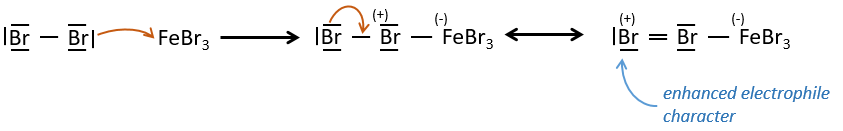
Ils améliorent le caractère électrophile de l’halogène qui peut désormais être attaqué par le substrat aromatique.
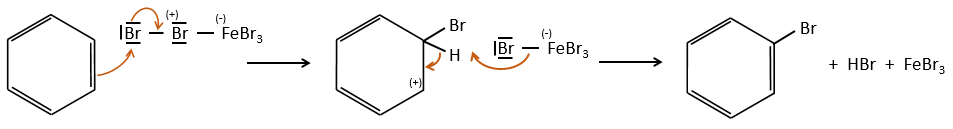
FeBr4– agit comme une base pour prendre un proton et fermer l’anneau.
Dans le tableau suivant nous pouvons trouver des énergies de liaison impliquées dans le processus pour les halogènes. Sur la gauche nous avons les énergies pour les réactifs et dans la colonne du milieu nous avons les énergies pour les produits. L’enthalpie de réaction est représentée sur la colonne de droite.
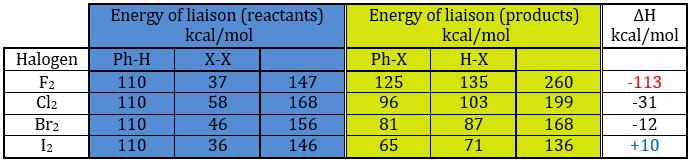
Si nous jetons un coup d’oeil à la variation de l’énergie impliquée par une halogénation, nous voyons que l’enthalpie de réaction est positive pour l’iode. La réaction n’est donc pas faite. La réaction est très exothermique pour F2. En fait cette réaction est explosive, Pour Cl et Br, nous avons besoin d’utiliser des catalyseurs (les acides de Lewis).
La sulfonation et la nitration :
SO3 et NO2+ sont suffisamment électrophiles pour se lier à un cycle aromatique.
La nitration :
Le nitrate peut être réduit de façon sélective pour obtenir une amine
Sulfonation :
L’atome de soufre est électrophile en raison du caractère électrocapteur des oxygènes. Cependant la réaction est réversible en présence d’eau pour former de l’acide sulfurique. Ce processus est exothermique et on devrait garder un oeil sur lui.
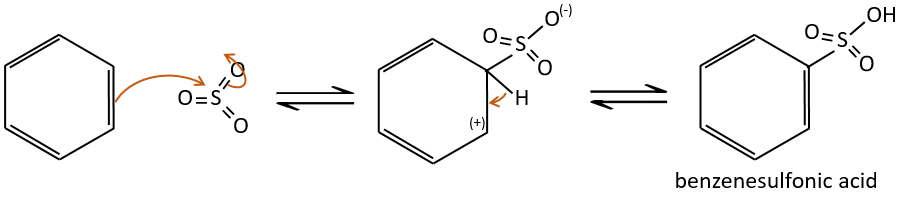
Nous produisons des détergents à partir de l’acide benzènesulfonique.
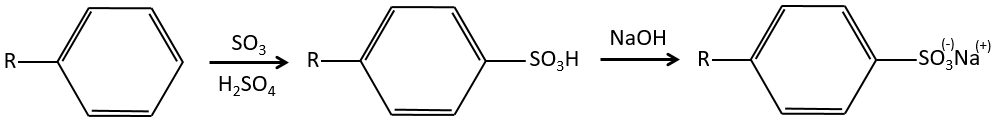
Et à partir de ce point, nous pouvons produire des sulfamides qui sont généralement de bons antibiotiques.
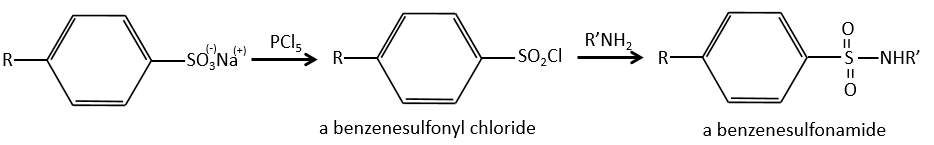
La première de ce genre était le Prontosil (4 – [(2,4-diaminophényl) azo] benzènesulfonamide) en 1932, développé par Domagk chez Bayer, qui a remporté un prix Nobel de médecine pour elle en 1939. Le programme de recherche a été conçu pour rechercher des agents qui pourraient agir comme médicaments antibactériens dans le corps. La découverte et le développement de ce premier sulfamide ont ouvert une nouvelle ère en médecine. Voici d’autres exemples d’agents antibactériens :
Alkylation de Friedel-Crafts :
Cette réaction, comme son nom l’indique, permet de lier une chaîne sur un cycle aromatique. La chaîne qui doit être ajoutée doit avoir un carbone électrophile. Encore une fois nous avons besoin d’un acide de Lewis pour faire cette réaction. La première étape est l’activation de l’halogénoalcane par l’acide de Lewis.
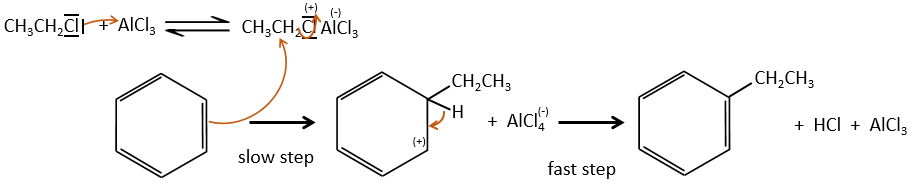
Ensuite le cycle attaque l’alcane, éjectant l’acide de Lewis halogène. Le produit de cette étape est appelé un intermédiaire de Wheland ou un ion arénium. Cette étape est l’étape de détermination de la réaction impliquant l’ouverture de la bague en raison de l’attaque électrophile. Un proton est prise par l’acide de Lewis halogéné pour donner l’aromaticité avant avec la récupération de l’acide de Lewis et la libération d’un acide. Cette étape est rapide par rapport à l’étape précédente. Comme résultat global nous avons ajouté une nouvelle chaîne de carbone sur le noyau phényle.
L’halogène sur la chaîne ajoutée n’est pas obligatoire. Un alcool ou d’autres précurseurs de carbocations peuvent aussi faire le travail.
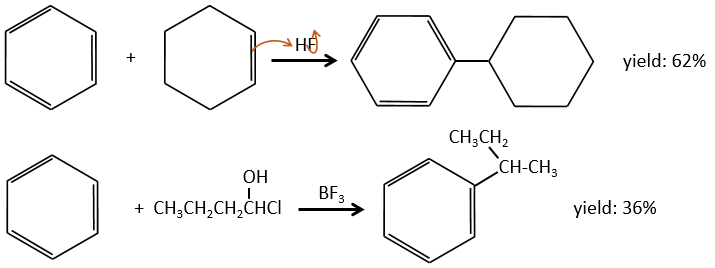
L’alkylation peut être intramoléculaire si elle aboutit à un nouveau cycle.
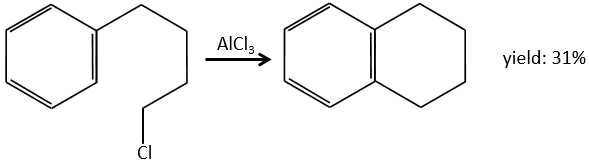
Les limitations de la méthode :
Le groupe alkyle qui a été ajouté au groupe phényle est un groupe donneur inductif. Cela signifie qu’il donne une partie de sa charge au phényle en augmentant sa capacité à attaquer les carbones électrophiles. En conséquence le processus d’alkylation ne cesse pas après l’ajout d’une chaîne si il y a encore de la place sur le phényle pour accueillir de nouvelles chaînes (nous verrons un peu plus tard sur quelle position, quel groupe peut être ajouté).
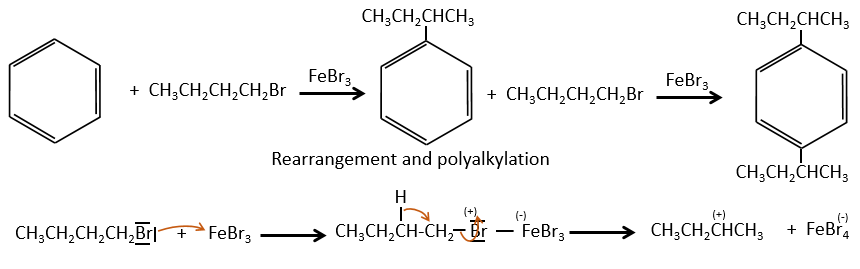
Une seconde limitation est le réarrangement du carbocation (comme d’habitude).
Acylation de Friedel-Crafts
La différence entre l’alkylation et l’acylation est que, pour ce dernier l’électrophile est un cation d’ acylium, conduisant à l’addition de -C = O-R sur le noyau phényle.
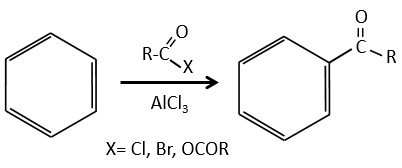
L’ion acylium est obtenu à l’aide d’un acide de Lewis
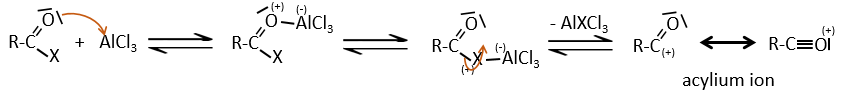
Le reste du processus est identique.
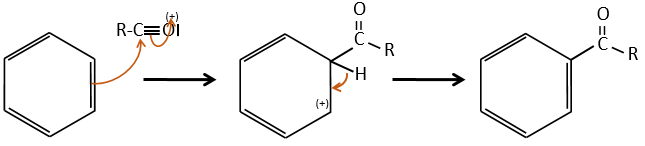
Cependant, il n’y a pas de polyacylation parce que le groupe carbonyle est un capteur inductif et mésomère en prenant des électrons du cycle. Il est possible de réduire la cétone avec la réaction de Clemmensen ou de Wolff-Kishner.
La régiosélectivité :
Un benzène qui porte déjà un groupe peut être attaqué sur 3 positions non équivalentes : ortho, méta et para. Certains groupes peuvent orienter la réaction vers les positions ortho ou para et d’autres groupes vers la position meta.
Effet mésomère :
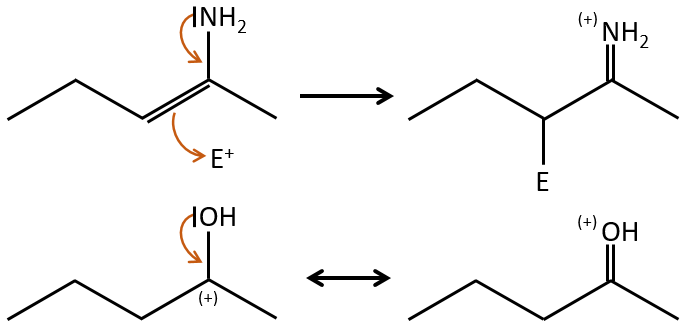
L’effet mésomère est la possibilité pour un hétéroatome de partager une paire solitaire avec des atomes voisins. Par exemple O et N sont des donateurs mésomères. Ils peuvent stabiliser les carbocations par le partage d’un de leurs paires seules qui contrebalancent la charge positive. Ils améliorent également le caractère nucléophile des liaisons doubles. Nous notons les donateurs mésomères par +M.
Les groupes peuvent également être accepteurs mésomères et sont notés -M. Par exemple, le groupe NO2 est un accepteur mésomère parce qu’il a une forme de résonance qui peut garder la charge supplémentaire.
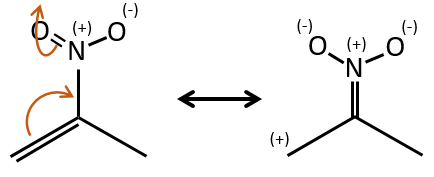
Notez que l’oxygène peut être mésomère donneur ou accepteur en fonction du groupe dans lequel il est et la structure de la molécule.
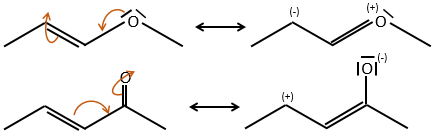
Effet inductif :
Lorsque deux atomes ont une électronégativité différente, il y a un déplacement d’électrons de l’atome moins électronégatif vers l’atome plus électronégatif. Les groupes ou atomes peuvent donc prendre ou donner des électrons depuis ou vers les atomes voisins. Les atomes ou groupes qui prennent les électrons du reste de la molécule sont des accepteurs inductifs et sont notées -I. Ils déstabilisent les carbocations et les charges positives et peuvent augmenter le caractère électrophile de l’atome avec lequel ils sont liés. Par exemple, le carbone d’un carbonyle donne une partie de sa charge à l’oxygène et porte une accusation partielle positif δ+. Un nucléophile est donc enclin à attaquer ce carbone.
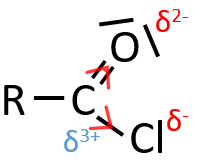
Les atomes qui sont moins électronégatifs que le carbone vont donner une partie de leur charge à la chaîne. Ceux-ci sont appelés les donneurs inductifs et sont notés +I. Le plus simple donneur inductif est H qui est moins électronégatif que le carbone. Cependant H est pris par convention comme l’intensité neutre de l’effet inductif. -CH3 est un donneur inductif parce que l’effet inductif de 3 hydrogènes est transmis à travers la chaîne carbonique (plus de 2-3 carbones). En conséquence les groupes alkyle sont des donneurs inductifs. L’effet est plus fort pour un groupe -C (CH3) 3 que pour -CH3 car le nombre d’atomes d’hydrogène qui peuvent partager leurs électrons est plus élevé.
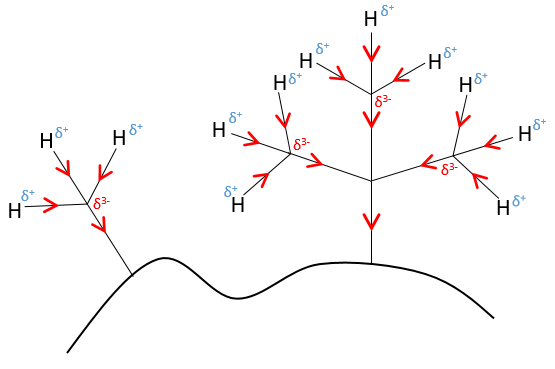
L’hybridation du carbone est une chose à prendre en compte pour déterminer l’effet inductif. Les électrons dans les orbitales « s » sont plus liés à l’atome que celles des orbitales p. En conséquence l’électronégativité des atomes de carbone sp2 est légèrement supérieure à l’électronégativité des atomes de carbone sp3.
Régiosélectivité en fonction des substituants des cycles aromatiques
Les effets mésomères et inductifs sont cumulatifs et un atome d’azote dans un groupe NH2 est simultanément un donneur mésomère et l’accepteur inductif (+IM). L’effet mésomère prévaut toujours en intensité.
Les donneurs inductifs :
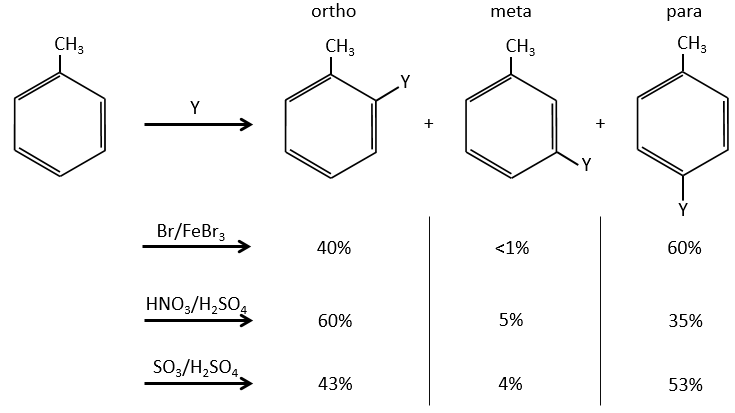
Les groupes qui sont des donneurs d’électrons par hyperconjugaison (ou les donneurs inductifs) activent ou favorisent l’ajout de nouveaux groupes sur le carbone. La substitution est orientée vers les positions ortho et para parce que le carbocation qui en a été fait au cours de la première étape de la substitution peut être stabilisée par le donneur inductif.
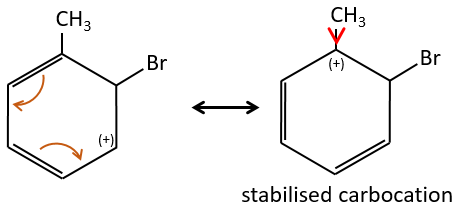
Il est impossible de placer la charge positive à l’endroit du groupe si la substitution devait être faite sur la position méta. Habituellement la position para est favorisée par rapport à la position ortho en raison de l’encombrement stérique. Cependant certains groupes, tels que NO2, semblent montrer plus d’intérêt à la position ortho.
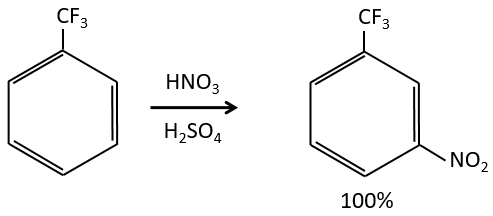
Les capteurs inductifs :
les électrocapteurs inductifs orientent la réaction dans la position méta. La raison est que dans les deux autres positions le carbocation peut être aux pieds du groupe CF3 qui veut prendre plus d’électrons, déstabilisant encore plus le carbocation.
Les donneurs mésomères :
les électrodoneurs mésomères activent et orientent la réaction en ortho/para parce qu’il y a une forme de résonance supplémentaire.
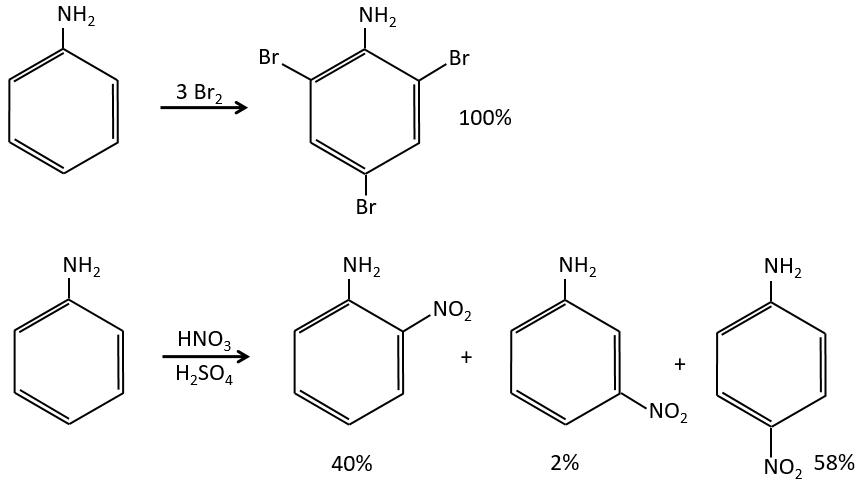
Notez que NH2, ainsi que l’oxygène de l’éther, est un donneur mésomère et un capteur inductif. L’effet mésomère est toujours plus important que l’effet inducteur.
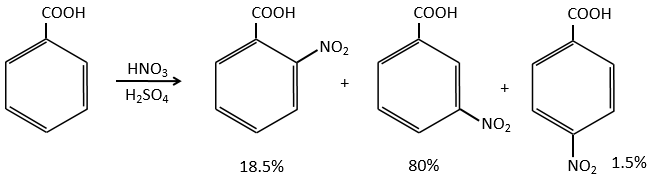
Les capteurs mésomères :
Les groupes qui sont électrocapteurs par résonance orientent la réaction dans la position méta.
Les halogènes :
les halogènes sont désactivants mais orientent la réaction en ortho et para.
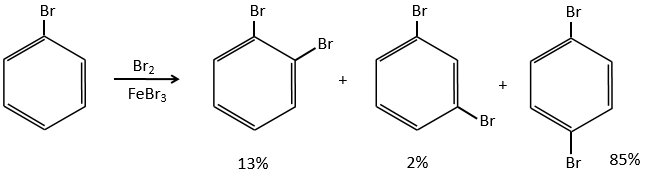
L’encombrement stérique ici est très important et explique la différence de population entre les réactions ortho et para.
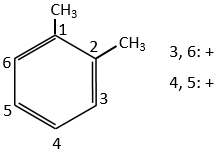
S’il y avait deux groupes sur le cycle phényle les effets sont additifs et la substitution est faite sur les positions les plus activées/moins désactivées.
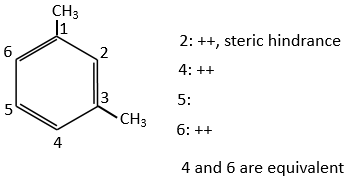
Les deux groupes méthyle du xylène (diméthylbenzène) indiqués ci-dessous sont activants et orientent dans ortho/para. Comme les deux sont des groupes d’activation ce réactif est plus réactif que le toluène. Les positions 2, 4 et 6 sont ainsi favorisées. Les positions 4 et 6 sont légèrement plus probables car il y a moins d’encombrement stérique. Dans le cas du xylène les fentes sont à peu près équivalentes.
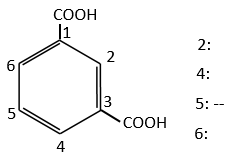
Dans le cas des groupes de désactivation la même réflexion est faite. Le COOH oriente en position méta et la position 5 est la position la moins désactivée.
La substitution nucléophile :
La substitution nucléophile sur des noyaux aromatiques est plus lente que la substitution électrophile et les substitutions sur des carbones sp3.
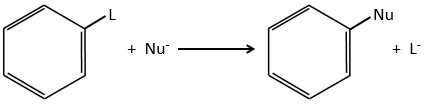
La raison en est que le cycle est déjà plein d’électrons. En outre un carbone sp2 est plus électronégatif que le carbone sp3. Il est donc difficile d’ajouter un nucléophile (qui aime les charges positives) sur un groupe phényle et la présence d’un groupe de capteur sur le phényle est nécessaire pour stabiliser le carbanion. Le groupe partant doit être un bon.
Plusieurs mécanismes sont possibles pour ajouter un nucléophile sur un substrat aromatique. L’un d’eux est l’addition-élimination.
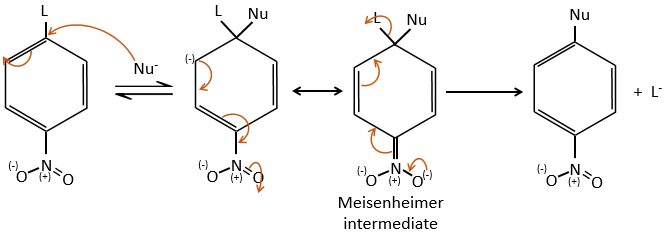
Le groupe d’activation (ici NO2) est nécessaire pour stabiliser la charge négative du carbanion formé lors de la première étape de la réaction qui est la plus lente. Le complexe intermédiaire est appelé complexe intermédiaire de Meisenheimer.
La séquence de réactivité halogène (comme groupe partant) est opposée à celle de SN2. Sur une chaîne aliphatique le clivage de la liaison C-X est faite au cours de l’étape déterminant de la SN2. Sur un cycle aromatique le clivage ne se produit pas au cours de l’étape déterminante. En outre les halogènes prennent des électrons du cycle et les petits halogènes sont donc plus réactifs que les grands. En conséquence nous avons les séquences de réactivité suivantes :
Aliphatique : F<<Cl<Br<I
Aromatique : F>>Cl>Br>I
Un atome hétérogène dans le cycle (O ou N) peut jouer le rôle du groupe de capteur.
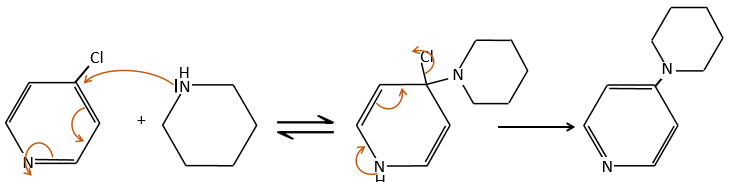
Une application de ce mécanisme est la détermination de l’acide aminé terminal des peptides. Le peptide réagit avec un composé aromatique par l’intermédiaire de son groupe amine terminal.
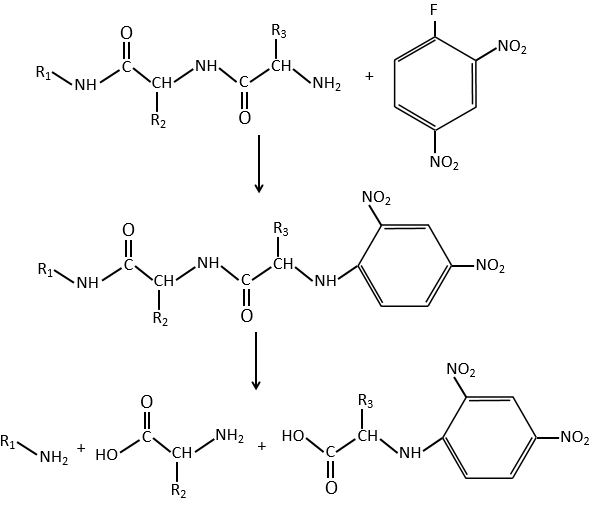
Une fois que les deux substrats sont liés ensemble nous hydrolysons les liaisons peptidiques (cf les amides). Tous les acides aminés sont maintenant séparés mais seulement l’un d’eux est lié au groupe aromatique.
Le mécanisme SN1
Ce mécanisme est surtout utilisé pour produire des sels d’arènediazonium. Cette espèce est obtenue à partir de l’aniline C6H7N avec NaNO2 dans un environnement acide.
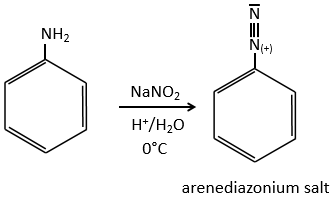
Le mécanisme est le suivant :
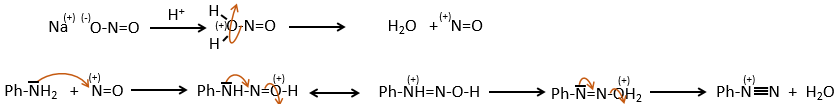
Les sels d’arènediazonium sont stables à basse température et perdent N2 à des températures plus élevées, ce qui libère un cation phényle qui peut réagir avec un nucléophile.
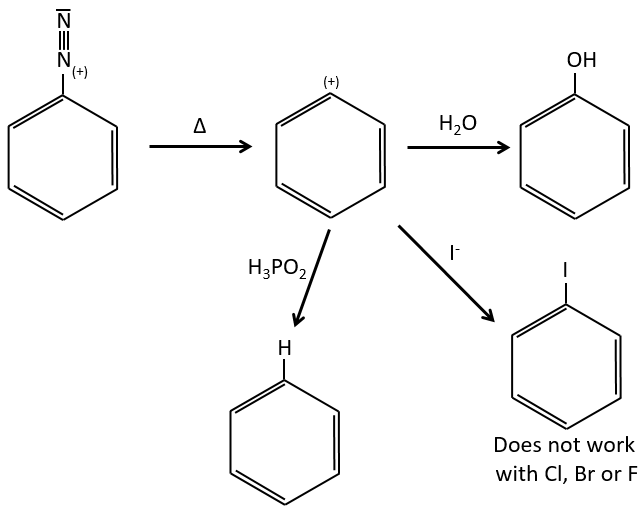
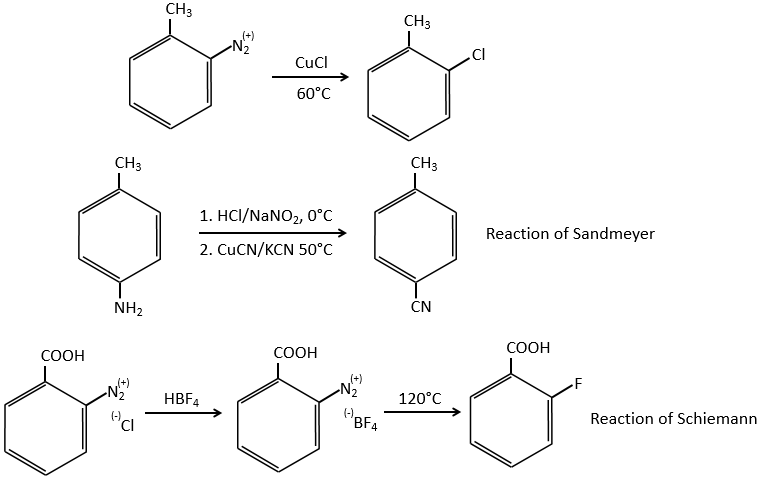
Les halogènes autres que l’iode ne donnent pas de bons résultats en raison de réactions secondaires. Pour ajouter Cl, Br ou F sur un cycle aromatique, on utilise la réaction de Sandmeyer en utilisant des sels de cuivre tels que CuCl, CuBr ou CuCN. Le mécanisme est un peu plus complexe et fait intervenir des radicaux.
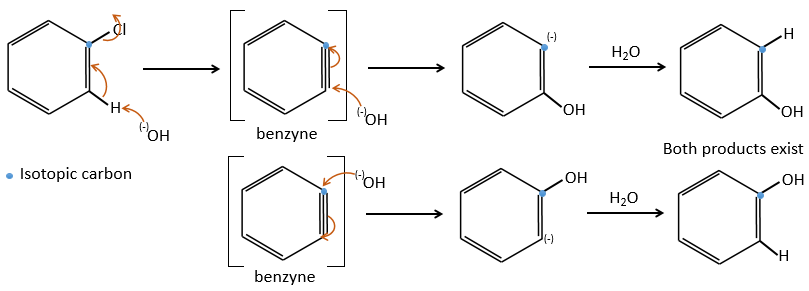
Le mécanisme comportant un benzyne :
Normalement les halogenoarenes ne peuvent pas faire des réactions SN2 ou SN1. Toutefois, dans des conditions très dures de température et de pH, il est possible de forcer pour faire de telles réactions.
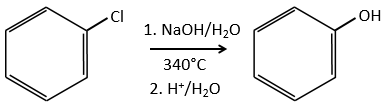
Le mécanisme implique une triple liaison dans le cycle, les espèces étant appelée benzyne, l’existence de cet intermédiaire a été montrée par un marquage isotopique. Le C lié à l’halogène est un isotope et nous observons un mélange racémique en tant que produit: