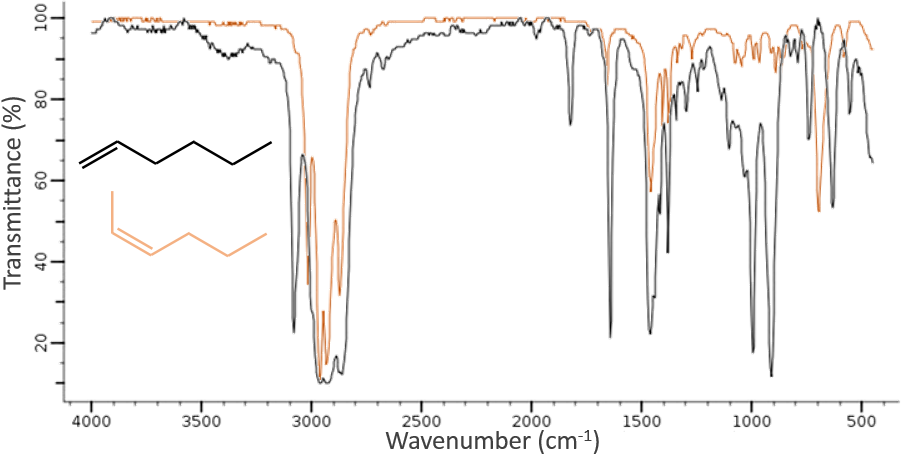
La spectroscopie infrarouge a un objectif différent de l’UV / visible. C’est un outil très puissant pour déterminer la structure des composés organiques. Un spectre IR est semblable à l’empreinte digitale d’une molécule et l’appariement des pics peuvent nous dire si une molécule est dans l’échantillon ou non. Le spectre montré ci-dessous est celui du 1-hexène en noir et du cis-2-hexène en orange. La différence de position de la double liaison conduit à une énorme différence dans le spectre IR.
La spectroscopie UV / visible implique des transitions électroniques tandis que la spectroscopie IR implique transitions entre états de vibration. Quant à l’UV / visible, on observe une série de bandes, et non des rayons, en raison des états de rotation subordonnés. Il existe deux principaux modes de vibration: l’allongement et la flexion.
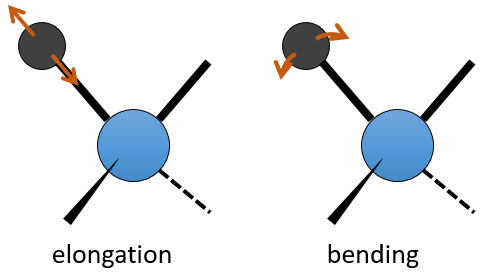
Une liaison entre deux atomes n’a pas une longueur constante. Les atomes vibrent autour de leur centre de masse avec une fréquence qui est caractéristique de la paire d’atomes. Il est le mode d’allongement: une vibration dans l’axe de la liaison. Cependant, les atomes sont souvent liés à plus d’un autre atome, en modifiant la fréquence de la vibration et donnant lieu à des processus d’allongement supplémentaires.
La flexion est une vibration hors de l’axe de la liaison. Elle conduit à une variation des angles entre les liaisons.
Pour une molécule de N atomes, le degré de liberté de la molécule est égale à la somme des degrés de liberté de chaque atome. Chaque atome a 3 degrés de liberté correspondant aux 3 coordonnées cartésiennes nécessaires pour décrire sa position dans la molécule. La molécule a donc 3n degrés de liberté. Parmi les 3n, 3 sont utilisés pour décrire les modes de translation et 3 sont utilisés pour décrire la rotation (2 si la molécule est linéaire). Il y a donc modes 3n-6 (ou 3n-5) modes de vibration pour chaque molécule. Par exemple, l’eau a 3 modes de vibration: 2 modes d’allongement et une mode de flexion.
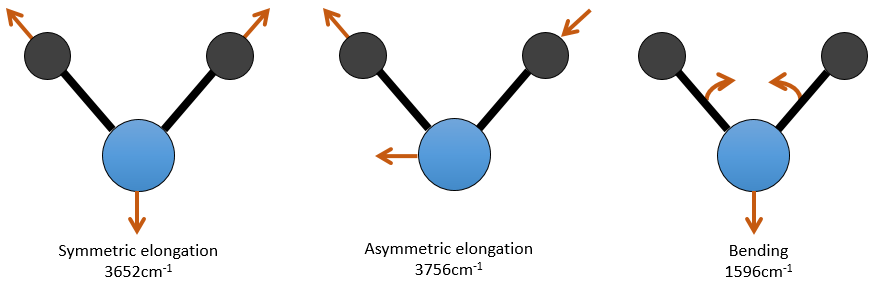
Un mode d’allongement est symétrique (le centre de la masse est décalé) et l’autre mode est asymétrique. Chaque mode représente une vibration spécifique, avec une longueur d’onde donnée. Les longueurs d’onde des modes d’allongement sont très proches les uns des autres et le mode de flexion a un nombre d’onde plus petit. Le fait que les modes d’élongation et les modes de flexion sont éloignés en termes de longueur d’onde et que les modes de flexion ont de plus petites longueurs d’onde sont des généralités que nous pouvons trouver pour toute liaison.
Il y a 3 atomes de carbone dans le dioxyde de carbone, mais 4 modes de vibrations comme le CO2 est linéaire (3n-5).
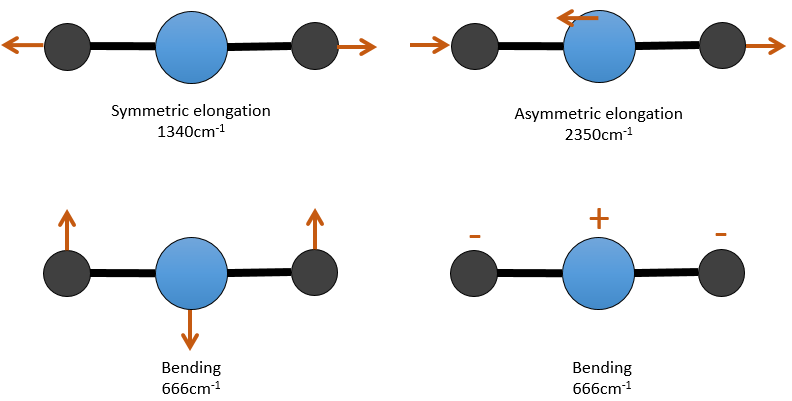
Il y a une forme d’allongement symétrique et une autre asymétrique et deux modes de flexion équivalentes (une dans le plan x-y et une dans la y-z). Les modes de flexion ont la même longueur d’onde à 666cm-1. Nous disons que ces modes de vibration sont dégénérées deux fois. En outre, l’allongement symétrique est inactive en IR parce que ce mode de vibration ne provoque aucune variation du moment dipolaire de la molécule.
Dans les grandes molécules, il est rare d’observer le nombre exact de modes de vibration (3n-6) parce que certaines modes viennent de la combinaison de deux ou plusieurs vibrations ou sont des harmoniques de puissants modes de vibration. Ces deux effets augmentent le nombre de bandes tandisque d’autres effets diminuent ce nombre par example:
– des bandes qui sont trop faibles pour être observés
– des bandes qui sont trop proche et qui fusionnent
– des bandes dégénérées
– des formes inactives de vibrations
– des modes avec des nombres d’ondes en dehors de la période analysée, généralement entre 4000 et 400 cm-1.
Il est possible de se rapprocher de la fréquence de vibration d’une liaison donnée avec la loi de Hooke qui considère la liaison comme un oscillateur harmonique simple entre deux masses M1 et M2 .
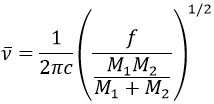
c est la vitesse de la lumière et f est une constante qui reflète la force de la liaison, dont la valeur est d’environ 5.105 dyne/cm (1 dyne =105N=105kg m s-2) pour les liaisons simples et deux et trois fois cette valeur pour les liaisons doubles et triples. f augmente de gauche à droite dans le tableau de Mendeleïev. Par exemple, la loi de Hooke donne une longueur d’onde de 3040cm-1 de la liaison C-H. Si on regarde les modes de vibration de CH2 à l’intérieur d’une chaîne carbonée, on trouve 6 modes de vibration (2 modes d’élongation et 4 modes de flexion).
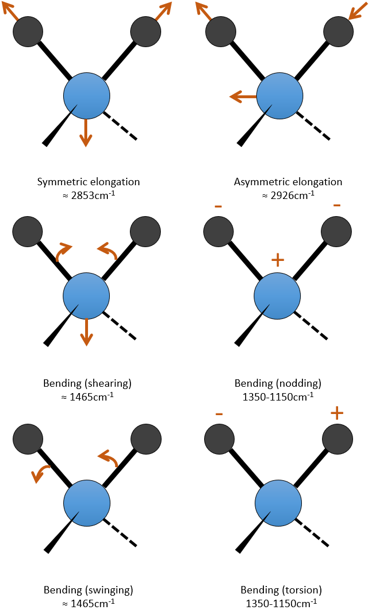
Rappelez-vous que la règle 3n-6 s’applique uniquement à des molécules complètes. Les modes d’allongement que nous observons ont des fréquences qui sont un peu inférieurs à ceux obtenus par la loi de Hooke (2926 et 2853cm-1 vs 3040cm-1) en raison de l’environnement de la liaison C-H qui ne sont pas pris en compte par Hooke.
De la loi de Hooke, il est évident que des atomes lourds vibrent à des fréquences plus petites. On peut à peine distinguer un spectre IR dans plusieurs régions en fonction de la paire d’atomes participant à la liaison et du type de liaison:
| 3800-2700cm-1: C-H, O-H, N-H | 1900-1500cm-1: C=C, C=O, C=N, N=O |
| 2300-2000cm-1: C≡C, C≡N | 1300-800cm-1: C-C, C-O, C-N |
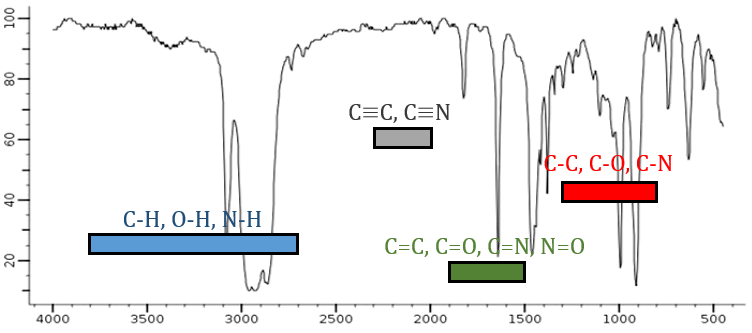
Sur la figure, nous pouvons donc en déduire déjà qu’il n’y a pas de liaison triple (région grise) dans notre molécule (le 1-hexène à partir de début), qu’il y a des liaisons doubles (région verte) et des liaisons simples (régions de bleu et rouge) . Cependant, nous ne pouvons pas encore déterminer quel pic correspond à laquelle les vibrations.
Interactions couplées
Lorsque deux oscillateurs partagent un atome commun, ils agissent rarement comme un des oscillateurs simples sauf si leurs modes de vibrations sont très différents. Le couplage entre deux modes de vibration produit deux nouveaux modes de vibration à des fréquences de plus en plus petite que celle en absence d’interaction.
Par exemple, dans le CO2, l’allongement asymétrique (2350cm-1) et l’allongement symétrique (1340cm-1) sont le résultat d’un couplage entre les deux oscillations C = O, l’ une raccourcit lorsque l’autre une allonge.
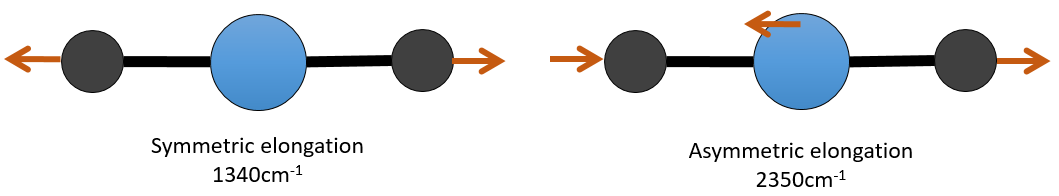
De ce que nous avons vu précédemment, un C = O devrait vibrer entre 1900-1500 cm-1, mais ce n’est clairement pas le cas ici. Le couplage entre les deux vibrations C = O a pour effet de déplacer la vibration vers les grandes fréquences (seul l’asymétrique est visible). Le couplage devient négligeable quand un ou plusieurs atomes de carbone séparent les agents de liaison. Deux carbonyles séparés par un ou plusieurs atomes de carbone montrerait une absorption vers 1725cm-1.
par conséquant pour avoir un couplage il faudrait que les vibrations partagent un atome et qu’elles aient des fréquences similaires. Cependant des interactions sont possibles entre les vibrations fondamentales et les vibrations harmoniques et/ou les vibrations de combinaison. Un tel couplage est appelé une résonance de Fermi. En fin pour le CO2, l’allongement symétrique n’est pas activé dans IR mais qui peut être observé dans le spectre Raman à 1340cm-1. En fait, il existe deux bandes à 1286cm-1 et 1388cm-1 en raison du couplage de la vibration avec le harmonique des modes de flexion (666cm-1). La première harmonique est donc à 1332cm-1 et interagit avec le mode d’allongement symétrique à 1340cm-1.
Des liaisons hydrogène
Les liaisons hydrogène diminuent les fréquences des liaisons et généralement élargit et augmente leurs bandes. L’importance de cet effet dépend de la force de la liaison H. Une liaison H est solide quand elle est dans la direction exacte de la paire libre de l’atome électronégatif. La force de la liaison dépend également de la distance entre les atomes, de l’atome et le fait que si un cycle peut être formé par la liaison H.
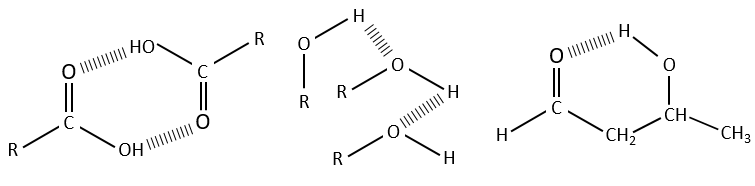
La diminution de la fréquence varie de 300 à plus de 500 cm-1 si la liaison H est intermoléculaire et est de moins de 100 cm-1 à plus de 300cm-1, si la liaison H est intramoléculaire. Les liaisons H sont souvent présentes entre une molécule et le solvant. Il est donc important d’indiquer le solvant et la concentration du composé sur un spectre. La présence d’eau dans l’échantillon a un effet visible sur les spectres.
Analysis of a spectrum
Un spectre IR montre la transmittance en fonction de la longueur d’onde / nombre d’ondes. Le spectre est à lire de haut en bas avec des bande descendant bas dans le spectre.
Une analyse précise d’un spectre IR n’est pas concevable. Les couplages, l’absence de modes de vibration et de la largeur de certaines bandes,rendent la détermination de la nature exacte des bandes doifficile. Cependant, comme il a été dit précédemment, un spectre IR est comme l’empreinte digitale d’un composé et si le spectre se superpose avec le spectre d’une molécule connue (avec le même solvant, la même concentration et la même configuration), le travail est fait. Dans d’autres situations, un spectre IR est utilisé en combinaison avec MS, UV et RMN.
Le spectre, habituellement entre 4000 et 400 cm-1 peut être divisé en deux régions importantes pour analyser: une entre 4000 et 1300 cm-1, appelées zone des groupes fonctionnels, et une sous-1 900 cm qui montre les modes de flexion.
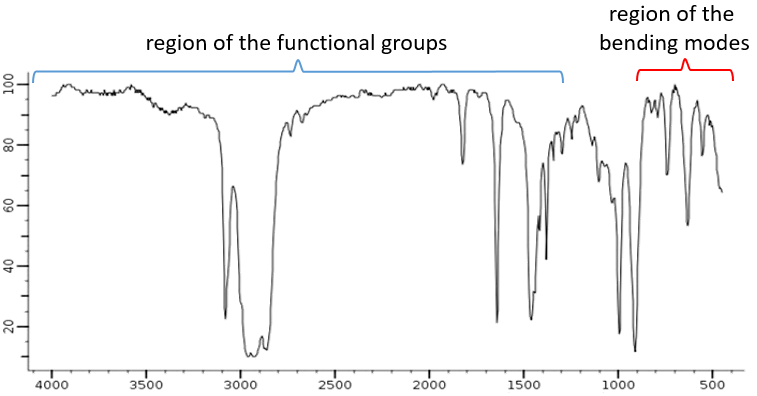
Les modes d’allongement de groupes fonctionnels avec OH, NH, C = O se trouvent dans la zone des groupes fonctionnels. Si cette région est vide, nous pouvons supposer que la molécule ne porte pas l’un d’eux. Dans de rares cas, une bande faible et/ou très large peut être confondue avec une absence de bande. Et encore il faut faire attention aux bandes de faible intensité qui peuvent aussi être des harmoniques ou des bandes de combinaison.
Les absorbances caractéristiques :
Il serait trop long de décrire toutes les bandes qui peuvent être observés sur un spectre IR. Le but ici est de montrer les bandes très caractéristiques qui peuvent être facilement repérés sur le spectre et d’avoir une idée générale de ce qui est dans la molécule et ce qui n’est pas. En combinaison avec un spectre de masse, il sera possible de déterminer la structure correcte de la molécule analysée.
Vibrations de hautes fréquences :
La région du spectre ci-dessus 2850 cm-1 est riche en informations qui peuvent facilement être étudié. Dans cette région, nous trouvons les bandes d’allongement de Y-H, Y = C, O, N. Les bandes sont généralement intenses et ne sont pas cachées, même si une autre bande est à proximité. La position d’une bande dépend de l’environnement direct de la liaison. Lorsque cela est nécessaire, nous écrivons cette liaison (simple, double ou triple) pour démarquer = C-H, à savoir une liaison hydrogène à un carbone avec une double liaison, du -C-H par exemple, à savoir une liaison hydrogène à un carbone avec une liaison simple.
Une première indication sur la nature du composé se trouve dans le voisinage de 3000 cm-1 où les liaisons C-H absorbent. La différence de fréquence entre aromatique = C-H et -C-H aliphatiques est petite mais est caractéristique:
Les C-H méthylique se trouvent en 2 bandes à 2962cm-1 et 2872cm-1 et les C-H méthylènique se trouvent un peu plus bas (2926 et 2853cm-1). Les = C-H se trouvent légèrement au-dessus 3000cm-1. Il est souvent synonyme d’un aromatique, mais il peut aussi être simplement un alcène (ou doubles liaisons conjuguées). Sur le spectre de l’hexène, le pic à 3080cm-1 est caractéristique de la double liaison et les pics pour le C-H sont cachés dans le grand groupe. Trois petits pics sont toutefois visibles autour des valeurs citées ci-dessus pour des -C-H méthylique méthylènique.
Il est donc facile de deviner la présence d’un cycle aromatique à partir des bandes -C-H mais il n’est pas facile de déterminer si la bande d’absorbance = C-H provient d’un cycle aromatique ou d’un alcène: car aussi bien le = C-H alcène que le =C-H aromatique montrent une bande intense dans la région des basses fréquences, respectivement entre 1000-650cm-1 et 900 à 675 cm-1. Les composés aromatiques (et hétéroaromatiques) vont également montrer 3 pics autour de 1600cm-1, 1500-1400cm-1 et 1300-1000cm-1(à noter que les alcènes conjugués montrent également ce genre de bandes). Toujours dans le spectre de l’hexène, le pic entre 1300-1000cm-1 n’est pas présent et le pic de flexion est inférieure à 675cm-1.
Les liaisons C-H des aldéhydes vibrent à des petits nombres d’onde, entre 2830 et 2695cm-1 parce que le carbonyle prend les électrons de la C-H et que cette liaison devient plus long, ce qui fait qu’elle vibre plus lentement. Le pic n’est pas très intense et peut être doublée en raison d’une résonance de Fermi avec la première harmonique du mode de flexion à 1390cm-1.
Les liaison ≡C-H absorbent à des fréquences supérieures à 3100cm-1, entre 3333cm-1 et 3267cm-1 mais ne sont pas les seules espèces absorbantes au dessus de 3100cm-1. Nous pouvons aussi trouver -O-H et les bandes d’élongation N-H ainsi que des liaisons hydrogène. Les bandes de ≡CH sont généralement plus minces que les bandes pour -O-H et -N-H et sont intenses.
Les Bandes d’allongement -O-H
Les bandes -O-H sont grandes et intense, et très grand dans le cas des acides carboxyliques. En effet, ces grandes bandes sont le résultat des liaisons H et la formation de dimères, lorsque la concentration d’alcool / acide sont bien dilué.
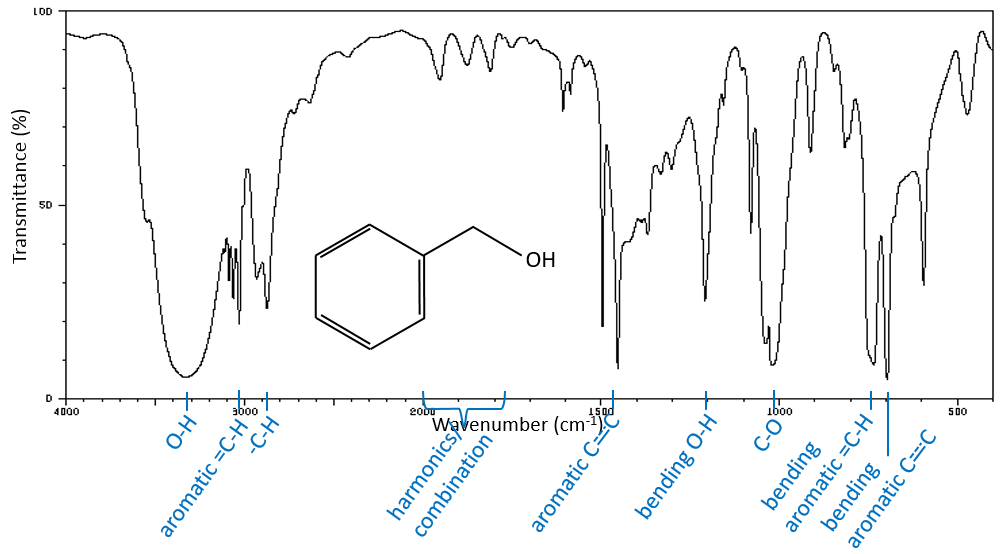
Les -O-H libres donnent des bandes petites et minces comprises entre 3650 et 3550cm-1 pour les alcools et autour de 3550cm-1 pour les acides, mais sont facilement effacées par les larges bandes de H: O-H qui se répandent sur des centaines de cm-1 sont de façon plus intense. Ces grandes bandes doivent être centrées entre 3550 et 3200cm-1(pour l’alcool benzylique, il est à 3320cm-1) pour les alcools et 3000cm-1pour les acides.
Les bandes d’allongement -N-H
Les bandes de fréquences élevées pour les amines aliphatiques sont assez faibles entre 3400 et 3250 cm-1. Il y a deux bandes dans le cas des amines primaires et une bande dans le cas d’une amine secondaire. Les modes d’élongation N-H sont légèrement déplacées vers les grandes fréquences si l’amine est aromatique. Le spectre est différente dans le cas des sels d’amine. Les ions ammonium donnent une bande d’absorption grande et intense entre 3300 et 3030cm-1. La fréquence diminue avec le degré de l’amine: amine primaire: 3000-2800cm-1, amine secondaire: 3000-2700cm-1 et amine tertiaire: 2700-2250cm-1).
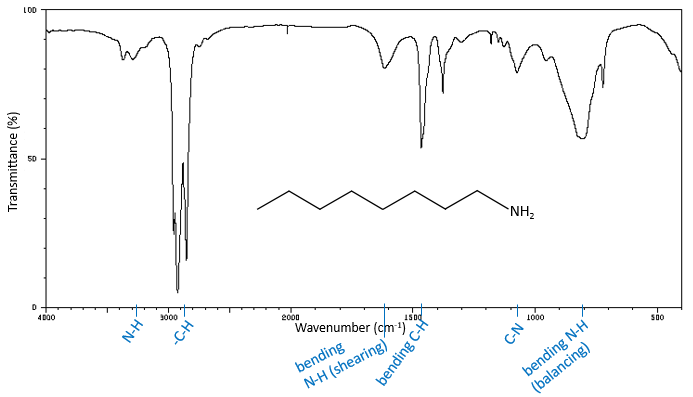
En l’absence de H liaison, l’amide primaire présente deux bandes autour de 3520 et 3400cm-1, tandis que les amides secondaires montrent une bande d’absorbance entre 3500 et 3400cm-1. En plus des solutions ou dans les solides concentré, liaisons H déplacent les bandes vers les petites fréquences, respectivement autour de 3350 et 3180cm-1 pour les amides primaires et plusieurs bandes entre 3330 et 3060cm-1 pour les amides secondaires. La présence de plusieurs bandes au lieu d’une seule bande est expliqué par la formation d’un variateur de lumière d’amides.
fréquences de milieu de gamme
Entre 2850 et 900 cm-1, nous pouvons trouver des bandes caractéristiques des vibrations d’allongement entre deux atomes autres que H et quelques vibrations de flexion de Y-H qui peuvent confirmer les observations de la région des hautes fréquences du spectre IR. Nous allons nous concentrer sur les bandes d’élongation.
Les liaisons S-H produisent une petite bande d’absorption entre 2600 et 2550cm-1. Cette bande est solitaire dans cette région du spectre, mais dans des solutions diluées de la bande peut être trop petite pour être détectée.
C≡C montre une petite bande d’absorbance entre 2260 et 2100cm-1. Cette région est dédiée à liaisons triples (C≡N montre une bande entre 2260 et 2240cm-1), mais les harmoniques et les bandes de combinaison peut être trouvée dans cette région aussi. Les bandes de C = C sont ou d’intensité moyenne ou faible entre 1667 et 1640cm-1 pour alcènes qui ne sont pas conjugués. Ces alcènes se trouvent entre 1650 et 1600 cm-1. Les alcènes cumulés (c = c = c) absorbent entre 2000 et 1900cm-1.
vibrations de C Allongement = O
L’absence d’absorbance entre 1870 et 1540cm-1 est synonyme de l’absence de carbonyle. Dans cette région, la position du pic dépend de la conjugaison du groupe carbonyle et des substituants (qui substituant et sa masse) et les liaisons possibles H.
Une cétone aliphatique se trouve à 1715cm-1. Modifications de l’environnement de la cétone peut induire soit une augmentation d’une diminution du nombre d’onde du pic qui dépend de la prédominance de l’induction ou de l’effet de résonance. L’effet inductif réduit la longueur de la liaison C = O, ce qui conduit à l’augmentation de sa fréquence de vibration. Une résonance augmente la longueur de la liaison C = O, entraînant le déplacement du pic vers le sommet C-O qui est à une fréquence plus basse. Par exemple, Cl a un effet inductif prédominant et le pic de la C = O est à 1815-1785cm-1. Le groupe carbonyle d’un amide primaire vibre à 1695-1650cm-1 parce que l’effet de résonance est prédominante. Il y a deux bandes pour les amides primaires et une seule pour les autres. Carbonyles conjugués avec des alcènes ont des pics d’absorption à plus petits nombres d’onde que la cétone aliphatique entre 1685 et 1666cm-1. Un aldéhyde absorbe un peu plus élevé que la cétone (1740-1720cm-1). Un acide absorbe fortement (de façon plus intense qu’une cétone) autour de 1760cm-1, mais des liaisons H dans les gradateurs d’acides peut déplacer le pic vers les petites fréquences (1720-1706cm-1). Esters absorbent à 1750-1735cm-1. Un halogénure d’acyle présente une forte absorption entre 1815 et 1785cm-1.
H liaisons diminuent la fréquence de vibration des carbonyles. L’effet est faible pour les obligations intermoléculaires H (une dizaine de cm-1), mais peut être important pour les obligations H intramoléculaires.
vibrations Allongement de C-O
Dans le spectre de milieu de gamme, entre 1300 et 900 cm-1 du spectre est généralement compliqué. Il est le « impression numérique » du composé où nous pouvons trouver les bandes d’alcools C-C-O qui devrait être accompagné de bandes O-H à grandes fréquences.
La liaison C-O des alcools présente une bande intense à cause des vibrations de l’allongement entre 1260 et 1000cm-1. Ce mode de vibration est généralement couplée à la prochaine carbone, à titre d’élongation C-C-O asymétrique. Le COC des éthers et des systèmes de époxydes peuvent également être trouvés dans cette région du spectre, entre 1150 et 1086cm-1 (pour son allongement asymétrique, symétrique étant très faible, car il ne modifie pas le moment dipolaire) comme une bande intense ou plusieurs bandes si les carbones à proximité sont ramifiées. Si un côté est un groupe aryle, on observe une bande intense (allongement asymétrique) à 1275-1200cm-1 et une bande intense (allongement symétrique) à 1075-1020cm-1). Enfin, les éthers de vinyle présentent des groupes similaires (1225-1200cm-1 et 1075-1020cm-1).
Les acides donnent deux bandes d’intensités inégales dans, en plus de celui de C = O provenant d’une interaction entre le mode d’allongement de C-O et le mode de flexion d’O-H. Nous parlons des bandes de C-O-H. La bande la plus intense entre 1315 et 1280cm-1 est appelée la bande d’élongation C-O. La deuxième bande, entre 1440 et 1395cm-1 est le groupe appelé la bande de flexion O-H. Cette bande tombe dans la même région que le mode de CH2 de cisaillement. Le carboxylate donne également deux bandes d’intensités inégales, la plus intense une entre 1650 et 1550cm-1 et l’autre autour de 1400cm-1.
Une particularité des esters est leur bande intense d’absorption à la place du mode de fonctionnement des cétones d’allongement. Cette bande est pas le groupe C = O, qui est déplacée vers les grandes fréquences mais l’une des deux bandes de C-O. Le groupe C = O est en effet entre 1750 et 1735cm-1 et 20cm-1 inférieur si le groupe carbonyle est conjugué. La vibration C-O est couplé avec les deux côtés de la liaison, ce qui conduit à deux bandes d’absorption. Le plus intense est une C-C (= O) -O donnant un pic entre 1300 et 1000cm-1. Les esters d’acides aromatiques absorbent fortement entre 1310 et 1250cm-1. L’autre bande entre 1111 et 1031cm-1 provient du couplage O-C-C. Sa fréquence dépend du substituant de l’ester. Esters d’alcools primaires sont les fréquences les plus basses (1064-1031cm-1), tandis que les esters d’alcools secondaires et d’aromatiques absorbent autour de-1et 1100cm 1111cm-1 respectivement.
vibrations Allongement de NO
groupes nitro ont un symétrique et un mode asymétrique d’élongation de la vibration. Le mode asymétrique donne une bande intense entre 1661 et 1499cm-1. Le mode symétrique donne une bande comprise entre 1389 et 1259cm-1. Les nombres d’ondes des deux bandes dépendent du substituant du groupe nitro. Dans le cas d’un alcane, les bandes doivent être observées autour de 1550 et 1372cm-1, respectivement. Un groupe électronégatif sur le carbone alpha augmente la fréquence tandis qu’une conjugaison diminue la fréquence.
Nitrates montrent 2 bandes intenses d’allongement pour N = O (une asymétrique et une symétrique) entre 1300-1255cm-1 et 1660-1625cm-1. Les nitrites ont deux bandes ainsi, l’un pour les cis (1625-1610cm-1) et un pour l’isomère trans (1680-1650cm-1).
composés nitroso montrent une bande d’absorption entre 1585 et 1539cm-1.
modes de C = S Allongement
Les modes d’allongement de C-S sont présents dans les basses fréquences aller et C = S légèrement au-dessus de cette région, entre 1250 et 1020cm-1. Cette bande est moins intense que celle d’un groupe C = O, car il est moins polaire.
modes de S = O Allongement
Les liaisons S = O produisent des bandes intenses en général. La liaison S = O de sulfoxydes vibre entre 1070 et 1030cm-1. Sulfones montrent deux bandes d’absorption entre 1350-1300cm-1 et 1160-1120cm-1. Les bandes sont décalées vers des fréquences plus importantes dans le cas d’un chlorure de sulfonyle (1410-1380cm-1 et 1204-1177cm-1) ou d’un sulfonamide (1370-1335cm-1 et 1170-1155cm-1).
Pliage dans la région de milieu de gamme
Plusieurs liaisons avec les modes d’allongement de la haute fréquence, typiquement Y-H, les liaisons ont modes de flexion qui tombent dans la même région que d’autres modes d’allongement (Y-Z). Ils compliquent la lecture du spectre, mais peuvent également confirmer des pics de la région des hautes fréquences.
C-H modes de flexion
Les modes de flexion des liaisons méthyléniques C-H ont été vu au début de ce chapitre. Un mode est dans la région basse des fréquences 720cm-1 et les trois autres sont dans la région de milieu de gamme. Le mode de cisaillement a une wavenumber presque fixe de 1465cm-1, mais les deux derniers modes (nod et torsion) donnent des pics entre 1350 et 1150cm-1. Dans un cycle, ces fréquences sont légèrement diminué. Méthyliques C-H ont deux modes de flexion: l’une où les trois H oscillent en phase et un dans lequel un H ne sont pas en phase. Le mode symétrique (en phase) donne un pic à 1375cm-1 et le mode asymétrique à 1450cm-1, à peu près à la même fréquence que le mode d’un groupe C-H méthylénique cisaillement. = C-H 1415cm à absorber alcènes-1 en raison d’un mode de cisaillement. Des hydrocarbures aromatiques ont un mode de flexion hors du plan qui donnent des pics intenses dans la région des basses fréquences et un mode de flexion dans le plan donnent des pics entre 1300 et 1000cm-1.
Pliage O-H
Pics d’alcools ne sont pas très caractéristique et se trouvent dans la région comprise entre 1420 et 1330cm-1. S’il y a un proton sur l’atome de carbone portant le groupe -OH (par exemple les alcools primaires et secondaires), il existe un couplage qui donne deux bandes autour 1420 et 1330cm-1. Les alcools tertiaires donnent un seul pic. Dans le cas des acides, il y a une interaction entre le mode d’allongement de C-O et le mode de flexion de l’O-H (décrit dans la section C-O modes d’allongement). Le mode de flexion C-O-H devrait être trouvé entre 1440cm-1 et 1395cm-1.
Pliage N-H
Les amines primaires ont un mode de flexion qui donne un pic de moyenne à haute intensité entre 1650cm-1 et 1580 cm-1. Le mode de flexion des amines secondaires est difficilement détectable à l’exception des amines aromatiques secondaires qui absorbent environ 1515cm-1. Dans le cas des amides, le mode de flexion N-H doit être observée par une bande comprise entre 1655 et 1590cm-1 avec une intensité façon plus petite que l’intensité du pic C = O. Comme C = O absorbe dans la même région, les pics de l’allongement C = O et de flexion N-H peuvent fusionner.
Les basses fréquences région
Dans cette région, on trouve principalement les modes de flexion, mais certains modes d’élongation sont également présents. À mon avis, cette région est moins intéressant que les régions de fréquences hautes et moyennes et est assez difficile à lire parce que plusieurs modes ont de grandes gammes de fréquences possibles qui se chevauchent avec d’autres modes. Les plupart des pics caractéristiques sont les pics pour les gradateurs et pour les aromatiques. Entre 900 et 650 cm-1, une bande large et intense caractéristique d’un variateur de lumière d’acide, un amide ou une amine. Dans la même région, l’absence de pics intenses indique que la molécule ne soit pas un groupe aromatique.
modes de flexion du CH
Le pic du mode de méthylénique -C-H balancer à 720cm-1 a une faible intensité ou non visible. Une bande très intense (généralement les plus intenses du spectre) est visible entre 1000 et 650 cm-1 en cas d’alcène est présent dans la molécule, provenant d’un mode de flexion hors du plan. Il est habituellement ce genre de flexion hors du plan qui donne des bandes intenses dans la région des basses fréquences. Un pic très intense peut donc être observée pour les aromatiques entre 900 et 650 cm-1. Les alcynes donnent un pic épais et intense à des fréquences plus basses, entre 700 et 610 cm-1.
D’autres modes de flexion
Le mode de flexion des alcools donne un pic d’épaisseur entre 769cm-1 et 650 cm-1 en plus des modes de flexion de la région des fréquences moyennes. Dimères d’acides donnent une très large bande (pas aussi large que la bande de hautes fréquences qui donnent les acides, mais il est caractéristique) d’intensité moyenne centrée près 920cm-1. Amides donnent également une épaisse bande d’intensité moyenne, mais entre 800 et 666cm-1, encore une fois en raison d’une flexion hors du plan. Cette courbure est plus forte et à des fréquences plus élevées pour les amines (909-666cm-1).
La flexion hors du plan des amines et des amides de générer une moyenne / bande intense respectivement entre 1 et 909-666cm 800-666cm-1. les groupes nitro semblent montrer un mode de flexion entre 763 et 690cm-1.
modes de Allongement de la région basse des fréquences
Une bande d’allongement de la N-O apparaît entre 870-833cm-1 à partir des groupes nitro, et entre 850-750cm-1 à partir nitrites. C-S absorbe entre 700 et 600 cm-1.
Enfin, les halogènes ont leurs modes de allongements dans les basses fréquences. Dans une chaîne aliphatique, C-Cl absorbe entre 850 et 550 cm-1, C-Br entre 690 et 515cm-1, C-I entre 600 et 500 cm-1 et C-F entre 1,400 et 730cm-1. La bande de C-F est forte et grande. Si le substituant est un groupe aromatique, les bandes sont déplacées vers les grandes fréquences, au-dessus 1000cm-1 dans le cas des chlorobenzènes.