Définition :
Les termes du génie génétique et l’ADN recombinant se réfèrent à des techniques dans lesquelles l’ADN peut être coupé, rejoint, sa séquence déterminée ou la séquence d’un segment modifié en fonction de l’utilisation prévue. Par exemple, un fragment d’ADN peut être isolé d’un organisme, joint à d’autres fragments d’ADN, et mis dans une bactérie ou d’autres organismes. Dans un autre exemple du génie génétique un fragment d’ADN, souvent un gène entier, peut être isolé et sa séquence nucléotidique déterminée ou sa séquence nucléotidique peut être altérée par des méthodes mutagènes in vitro. Ces activités connexes en génie génétique ont deux objectifs fondamentaux :
1) pour en apprendre davantage sur les œuvres de la nature.
2) trouver les moyens de faire usage de ces connaissances à des fins pratiques.
L’isolement du phage spécialisé de transduction qui portait les gènes de l’opéron lac a été une avancée particulièrement importante, car ces phages ont fourni un enrichissement de plus de 100 fois des gènes lac par rapport à l’ADN chromosomique. Il a également stimulé une grande variété d’études importantes qui ont favorisé grandement notre compréhension de la régulation des gènes ainsi que favorisé le développement de nombreuses techniques de génie génétique importantes. Maintenant le génie génétique permet les mêmes sortes d’études à effectuer sur un gène à partir de pratiquement tout organisme.
La deuxième raison majeure de l’intérêt du génie génétique est la synthèse économique de protéines, difficiles ou impossibles à purifier à partir de leurs sources naturelles. Ces protéines peuvent être des antigènes pour une utilisation dans l’immunisation, les enzymes pour une utilisation dans les procédés chimiques ou des protéines spécialisées à des fins thérapeutiques. Les séquences d’ADN cloné peuvent également être utilisées pour la détection des défauts chromosomiques et dans des études génétiques. Beaucoup de recherches en génie génétique ont également été réalisées dans les usines avec l’espoir d’améliorer les méthodes génétiques traditionnelles de modification des cultures. Un second objectif est l’introduction de la résistance aux herbicides dans les cultures souhaitées. Cela permettrait la pulvérisation contre les mauvaises herbes dans les champs même pendant la croissance au lieu de le faire avant la plantation. Le génie génétique de l’ADN implique généralement les étapes suivantes :
1) L’ADN pour l’étude doit être isolé et libéré des contaminants. Il doit être possible de couper cet ADN à des endroits spécifiques afin de produire des fragments contenant des gènes ou parties de gènes.
2) Ensuite il doit être possible de rejoindre les fragments d’ADN pour former des molécules d’ADN hybrides.
3) Les vecteurs doivent exister pour que les fragments assemblés puissent être introduits dans des cellules par le procédé appelé transformation.
4) Les vecteurs doivent avoir deux propriétés. Tout d’abord ils doivent fournir l’ADN autonome du vecteur pour la réplication dans les cellules et, d’autre part, ils doivent permettre une croissance sélective des seules cellules qui ont reçu les vecteurs.
Ce chapitre décrit les étapes fondamentales du génie génétique ainsi que la technique essentielle de la détermination de la séquence nucléotidique d’un fragment d’ADN
L’isolement de l’ADN :
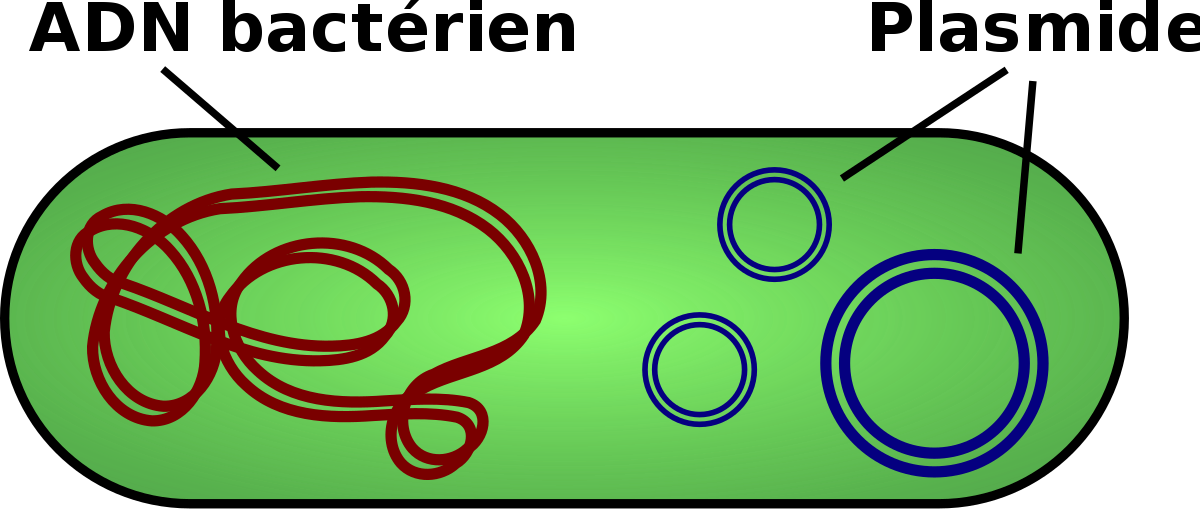
L’ADN cellulaire, chromosomique ou non chromosomique, est le point de départ de nombreux expériences de génie génétique. Un tel ADN peut être extrait et purifié par les techniques classiques de chauffage des extraits cellulaires en présence de détergents et après élimination des protéines par extraction au phénol. Si des polysaccharides ou des ARN contaminent l’échantillon ils peuvent être éliminés par centrifugation à gradient de densité d’équilibre au chlorure de césium. Deux types de vecteurs sont communément utilisés : des plasmides et des phages. Un plasmide est un élément d’ADN semblable à un épisome qui se reproduit de manière indépendante du chromosome. Habituellement les plasmides sont de petites tailles, 3000 à 25.000 paires de bases et circulaire.
En général, le phage lambda ou des dérivés apparentés sont utilisés comme vecteur pour des Escherichia coli, mais pour clonage dans d’autres bactéries, comme Bacillus subtilis, d’autres phages sont utilisés. Dans certains cas un plasmide peut être développé qui se réplique de façon autonome en dehors de plus d’un organisme hôte. Ces vecteurs « navettes » sont importants dans l’étude des gènes des eucaryotes, nous allons les examiner plus tard. Le plus souvent la purification complète de l’ADN de plasmide est inutile et l’ADN utilisable peut être obtenu simplement par la lyse des cellules en éliminant partiellement l’ADN chromosomique et la plupart des protéines. Les constructions complexes d’ADN nécessitent souvent l’ADN très pur pour éviter l’interférence des nucléases exogènes ou l’inhibition des enzymes sensibles.
L’isolement de l’ADN plasmidique :
La purification complète de l’ADN plasmidique nécessite généralement plusieurs étapes. Après que les cellules sont ouvertes avec le lysozyme qui digère la paroi cellulaire et que les détergents sont ajoutés pour solubiliser les membranes et inactiver certaines protéines, la plupart des ADN chromosomiques sont éliminés par centrifugation. Pour de nombreux objectifs des méthodes chromatographiques peuvent être utilisées pour effectuer la purification. Lorsque la pureté la plus élevée est nécessaire le plasmide est purifié par la densité d’équilibre de centrifugation à gradient. Cela se fait en présence de bromure d’éthidium. Tout ADN chromosomique restant avec le plasmide aurait été fragmenté et sera linéaire, alors que la plupart des ADN plasmidique seront circulaires car liés de manière covalente. Comme nous l’avons vu précédemment l’intercalation de bromure d’éthidium détord l’ADN.
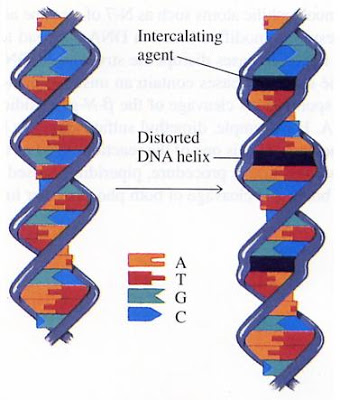
Pour une molécule circulaire cette détorsion génère sur-enroulement alors que pour une molécule linéaire la détorsion n’a pas d’effets majeurs. Par conséquent un ADN linéaire peut intercaler plus du bromure d’éthidium qu’une molécule circulaire. Etant donné que le bromure d’éthidium est moins dense que l’ADN, les molécules d’ADN linéaires intercalées avec bromure d’éthidium « flottent » par rapport à l’ADN circulaire et donc les deux espèces peuvent facilement être séparées. Suivant la centrifugation, les deux bandes d’ADN sont observées par les UV éclairant la lumière du tube. La fluorescence naturelle du bromure d’éthidium est augmentée de 50 fois par intercalation dans l’ADN et le Phage lambda peut également être partiellement purifié par des techniques rapides et éliminer les débris cellulaires et la plupart des contaminants. Une purification plus complète peut être obtenue en utilisant leur densité unique de 1,5 g/cm3, qui est à mi-chemin entre la masse volumique de protéines, de 1,3 et la densité de l’ADN, 1.7. Le phage peut être isolé par la centrifugation à gradient de densité d’équilibre dans laquelle la densité à mi-chemin entre le haut et le bas du tube de centrifugation est de 1,5. Eux aussi peuvent être facilement observés dans la centrifugeuse. Ils forment une bande bleue qui résulte de la dispersion préférentielle des longueurs d’onde plus courtes de la lumière connue sous le nom d’effet Tyndall. Ce même phénomène est la raison pour laquelle le ciel est bleu et les couchers de soleil sont rouges.
La biologie des enzymes de restriction :
Dans cette section, nous allons d’abord voir la biologie des enzymes de restriction et puis revenir à leur utilisation pour couper l’ADN. On a maintenant trouvé un grand nombre d’enzymes qui coupent l’ADN à des endroits spécifiques. Pour la plupart ces enzymes proviennent de bactéries. Elles sont appelées des enzymes de restriction parce que, dans les quelques cas qui ont été soigneusement étudiés, le clivage enzymatique de l’ADN fait partie du système de restriction-modification de la cellule. Le phénomène de restriction-modification des bactéries est un système immunitaire à une petite échelle pour une protection contre l’infection par l’ADN étranger. Contrairement à des organismes supérieurs dans lesquels l’identification et l’inactivation de l’invasion des parasites, des bactéries ou des virus peuvent être effectuées de manière extracellulaire, les bactéries peuvent se protéger après que l’ADN étranger est entré dans leur cytoplasme. Pour cette protection de nombreuses bactéries marquent spécifiquement leur propre ADN par la méthylation des bases sur des séquences particulières avec des enzymes de modification. L’ADN qui est reconnu comme étranger par l’absence de groupes méthyle sur ces mêmes séquences est clivé par les enzymes de restriction puis dégradé par les exonucléases aux nucléotides. Moins d’un phage sur 10000 sont erronément méthylés et sont capables de croître et de lyser un E.coli protégé par certains systèmes de modification-restriction. En outre les bactéries se protègent de l’ADN végétal et animal. L’ADN de beaucoup d’ animaux et de végétaux est méthylé sur la cytosine dans les séquences CpG. Beaucoup de souches de bactéries contiennent également des enzymes qui clivent l’ADN lorsqu’il est méthylé sur des positions spécifiques.
Arber a étudié la restriction du phage lambda dans E. coli et a constaté que La souche E. coli C ne contenait pas de système de modification-restriction. Souche B a un système de modification-restriction, et pourtant un autre reconnaît et méthyle une séquence nucléotidique différente dans la souche K-12. Phage P1 possède un système de modification- restriction propre à elle même qui est superposable au système de restriction-modification de l’hôte dans lequel elle est un lysogène.
Soit la notation λ-C représente la croissance de phage lambda qui a été cultivé sur La souche E. coli C. L’infection des souches B, K-12, et C avec λ sur diverses souches va se faire à différentes vitesses :
La possession d’un système de modification-restriction introduit des complexités dans le processus de replication de l’ADN. Imaginez que le double brin d’ADN contient des groupes méthyle sur les deux brins de l’ADN au niveau d’une séquence de reconnaissance. la réplication de l’ADN crée un duplex dans lequel l’un des brins des duplexes fille dans un premier temps n’a pas été modifié .
Cet ADN demi-méthylé ne doit pas être reconnu comme un ADN étranger et clivé, mais doit être reconnu comme «soi» et méthylé.
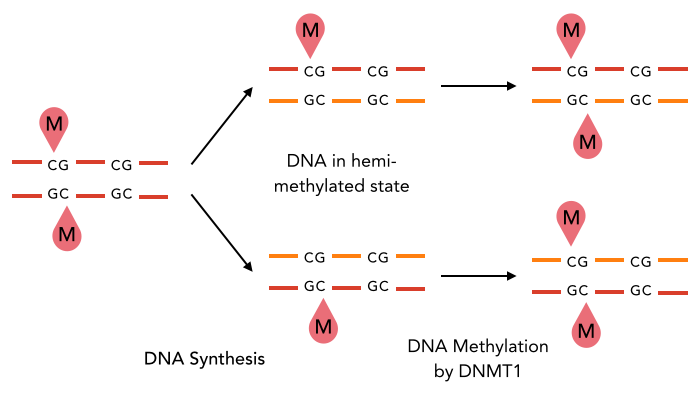
Par conséquent, le système de restriction- modification fonctionne comme un micro-ordinateur, reconnaissant les trois états différents de la séquence de reconnaissance il réagit de trois manières différentes :
1) Si la séquence n’ est pas méthylé, les enzymes la clivent.
2) Si l’ADN est méthylé sur l’un des deux brins, le système de modification méthyl l’autre brin.
3) si l’ADN est méthylé sur les deux brins, les enzymes ne font rien.
Une séquence de reconnaissance palindromique simplifie le fonctionnement du système de modification-restriction. Un palindrome est une séquence qui se lit de la meme manière de gauche à droite et de droite à gauche, comme les mots « REPAPER et RADAR ». Comme les brins d’ADN possèdent une direction, on considère une séquence d’ADN à palindrome si elle est identique à la lecture de 5 ‘à 3′ sur le brin supérieur et le brin inférieur . les Palindromes, peuvent être de toutes tailles, mais la plupart de ceux qui sont utilisés en tant que séquences de reconnaissance de modification-restriction sont quatre, cinq, six, et rarement, huit bases. En vertu des propriétés de palindromes, les deux filles de palindromes répliquées ont des séquences identiques, et donc l’enzyme de modification doit reconnaître et méthyler un seul type de substrat (Fig. 9.4).
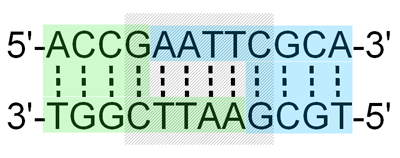
Comme nous l’avons déjà vu précédemment pour la reconnaissance des séquences non palindromic, il faudrait que l’enzyme de modification reconnaîsse à la fois les deux séquences différentes. On peut supposer que les protéines dimères sont utilisés pour les reconnaître. Les enzymes de restriction sont divisés en trois catégories principales. les enzymes
1) Classe I : ces enzymes sont formées de 3 sous-unités (d’une sous-unité de clivage,une sous-unité de méthylation et une sous-unité de reconnaissance des séquences). Ces enzymes clivent à
des sites éloignés de leurs séquences de reconnaissance et ne seront pas discuté ici même si elles ont été les premiers à etre découvertes.
2) Classe II :les enzymes de classe II possèdent leur sous unité de reconnaissance des séquences et leur sous unités de clivage ensemble. Ils coupent près de leur séquence de reconnaissance et sont le plus utilisé dans le génie génétique.
3) classe III possèdent 3 sous-unités (la sous-unité de clivage associée à celle de reconnaissance et la sous-unité de méthylation). Ces enzymes coupent près de leur site de reconnaissance.
Une enzyme de restriction dans une cellule est une bombe à retardement parce que les principes physicà-chimiques
limitent leur spécificité pour la liaison. Si une enzyme de restriction se liait à une séquence erronée, et comme une bactérie typique contient environ 4 × 1000000 de telles séquences, très probablement la séquence ne serait pas méthylé et l’enzyme pourrait cliver l’ADN et la cellule mourrerait. l’observation, cependant, est que les cellules contenant des enzymes de restriction ne meurent pas plus vite que les cellules sans enzymes de restriction. Comment, alors, est généré la spécificité extraordinairement grande des enzymes de restriction ?
La coupure d’ADN avec des enzymes de restriction :
Les enzymes de restriction fournissent un outil nécessaire pour couper des fragments de l’ADN à partir de molécules plus grandes. Leur spécificité permet une très grande sélectivité, et leur grand nombre ( plus d’une centaine de restrictions différentes), permet une trés grande variétés de choix des sites de clivage utilisés. Souvent, les fragments peuvent être produits avec des points d’extrémité situés à moins de 20 paires de bases de n’importe quel emplacement souhaité.
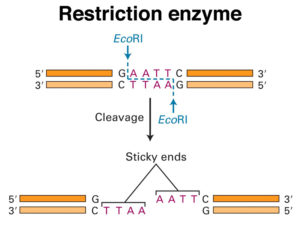
Une des propriétés les plus utiles des enzymes de restriction pour le génie génétique se trouve dans le système de modification-restriction produit par Plasmide R d’E.coli. L’enzyme de restriction correspondante est appelée Eco RI. Au lieu de cliver au centre de sa séquence de reconnaissance palindromique, cette enzyme coupe à l’extrémité et produit des extrémités à 4 bases complémentaires.Ces extrémités « collantes » sont les plus utiles dans la technologie de l’ADN recombinant. Elles peuvent être re-soudés à basse température comme les extrémités « collantes » du phage lambda. Cela permet l’assemblage efficace des fragments de l’ADN au cours des étapes de soudures . Environ la moitié des enzymes de restriction, actuellement connu peuvent générer des extrémités en surplomb ou collantes. Dans certaines situations, un fragment d’ADN peut même être agencé de telle manière à avoir deux types de collant différents.
L’isolement des fragments d’ADN :
Après le clivage de l’ADN par des enzymes de restriction ou d’autres manipulations (cf plus tard) les fragments d’ADN doivent souvent être isolés. Heureusement, le fractionnement en fonction de la taille est particulièrement facile parce que, comme indiqué précédemment, l’ADN possède un rapport charge sur masse constant et les fragments d’ADN double brin de la même longueur ont la même forme et donc migrent pendant l’électrophorèse à une vitesse pratiquement indépendant de leur séquence. En général, plus l’ADN est grand, plus sa vitesse de migration est lente. Une très grande résolution peut être obtenue en électrophorèse par example deux fragments dont les tailles diffèrent de 0,5% peuvent être séparés si elles se trouvent dans une plage de 2 à 50 000 paires de bases. Une plage typique pour une séparation de taille adéquate pourrait être de 5 à 200 paires de bases ou 50 à 1000 paires de bases, et ainsi de suite. Le matériau à travers lequel l’ADN est soumis à une électrophorèse doit posséder des propriétés particulières. Il doit être peu coûteux, facilement utilisable, non chargée, et il devrait former un réseau poreux. Deux matériaux répondent aux exigences: agarose et polyacrylamide. Après l’électrophorèse, les bandes formées par les différentes tailles des fragments peuvent être localisés par autoradiographie si l’ADN avait été radiomarqué avant la séparation. Habituellement 32PO4 est une étiquette commode parce que le phosphate se trouve dans l’ARN et l’ADN, 32P émet notamment des électrons énergétiques qui les rend facilement détectables, et enfin 32P a une demi-vie courte de sorte que la plupart des atomes radioactifs dans un échantillon désintégreront dans un délai raisonnable. L’isotope 33P est également utilisé. Sa désintégration bêta est plus faible, et il a une demi-vie de 90 jours. Souvent, l’ADN est suffisamment présent et il peut être directement détecté par coloration avec éthidium bromure. La fluorescence accrue du bromure d’éthidium intercalées dans l’ADN par rapport à sa fluorescence dans la solution permet la détection d’aussi peu que 5 ng d’ADN dans un groupe. Après la séparation par l’ électrophorèse et la détection de l’ADN, les fragments souhaités peuvent être isolé à partir du gel.
Souder les fragments d’ADN :
Après la coupure et la purification de l’ADN nous allons discuter de l’assemblage des molécules d’ADN. In vivo, l’enzyme ADN ligase répare les entailles survenant sur la squelette de l’ADN. Cette activité peut également être utilisés in vitro pour la jonction de deux molécules d’ADN. Deux exigences doivent être respectées. D’abord, les molécules doivent être les substrats corrects, autrement dit, ils doivent posséder des groupes hydroxyle 3′- et 5′-phosphate. Ensuite les groupes sur les molécules à assembler doivent être correctement positionnés par rapport à l’autre. Le procédé pour produire le bon positionnement présente deux variantes: soit hybrider les fragments ensemble par leurs extrémités collantes complémentaires :
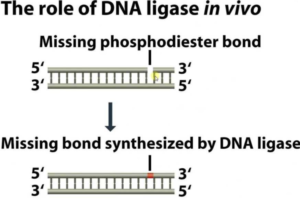
et, si l’assamblage concerne des fragments coupés avec bouts francs , d’utiliser des concentrations très élevées de fragments qui, de temps en temps,seront spontanément face à face avec positions correctes.De nombreuses enzymes de restriction telles que EcoRI produisent des extrémités collantes à quatre bases qui peuvent être soudées ensembles. Comme les extrémités collantes sont généralement seulement quatre paires de base, la baisse de la température à environ 12 ° C, facilite le processus d’hybridation-ligature. Les extrémités franches de molécules d’ADN qui sont générés par certaines enzymes de restriction génèrent des problèmes. Une solution consiste à convertir les bouts francs des molécules à des bouts collants par l’enzyme terminale transférase. Cette enzyme ajoute des nucléotides à l’extrémité 3′ de l’ADN. Une queue Poly-dG peut être ajouté à un fragment et une queue poly-dC peut être ajouté à l’autre et les fragments peuvent ensuite être hybridés ensemble
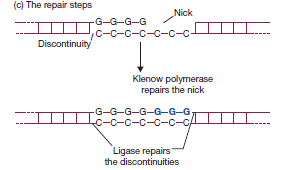
Si les queues sont assez longues, le complexe peut être introduit directement dans les cellules, où les lacunes et les pseudos seront remplis et scellés par le système enzymatique des cellules. Le plus souvent, la réaction en chaîne par polymerase ( PCR) serait utilisée pour générer des extrémités souhaitées sur les molécules. En outre les molécules ayant un bout franc peuvent également être soudées
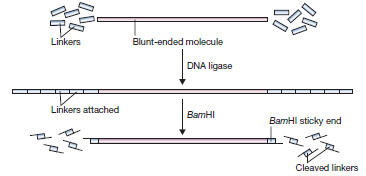
par l’ADN ligase. Bien que cette méthode est très simple, elle souffre de deux inconvénients: Elle nécessite des concentrations élevées d’ADN et de l’ADN ligase pour que la réaction s’initie et meme dans ces conditions l’efficacité reste faible. En outre, il est difficile d’exciser le fragment du vecteur . Les Lieurs peuvent également être utilisés pour générer des molécules auto-complémentaires monocaténaires. Les Linkers sont des ADN courtes avec bouts francs , contenant la séquence de reconnaissance d’une enzyme de restriction qui produit des extrémités cohésives. La ligature des lieurs à des fragments d’ADN se produit avec un rendement assez haut car des concentrations molaires élevées des lieurs peuvent facilement être obtenues. Une fois que les lieurs ont été relié au segment d’ADN, le mélange est digéré avec l’enzyme de restriction qui coupe les lieurs et génère des extrémités collantes. Ainsi une molécule d’ADN à bout franc est converti en une molécule collante à composition non limitée qui peuvent facilement être jointes à d’autres molécules d’ADN.