Vecteurs: la Sélection et réplication autonome de l’ ADN :
Le clonage d’un morceau d’ADN exige qu’il soit répliqué quand il est remis dans les cellules. D’où l’ADN à cloner doit lui-même être une unité répliquante indépendante, un réplicon ou doit être joint à un réplicon. De plus, étant donné que l’efficacité de l’introduction de l’ADN dans des cellules est bien inférieur à 100%, les cellules qui ont absorbé l’ADN et on dit qu’elles ont été transformées, doivent être facilement identifiables. En effet, étant donné que seulement une bactérie cellule sur 105 est transformée, les sélections doivent généralement être incluses pour autoriser uniquement les cellules transformées de croître. Les vecteurs doivent remplir les deux conditions décrites ci-dessus, la réplication dans la cellule hôte et la sélection des cellules ayant reçu l’ADN transformant. Comme mentionné précédemment deux principaux types de vecteurs sont utilisés : des plasmides et des phages. Les plasmides contiennent des réplicons bactériens qui peuvent coexister avec l’ADN cellulaire normal et au moins un gène sélectionnable. Habituellement c’est un gène conférant une résistance à une antibiotique. Les phages contiennent bien sûr des gènes pour la réplication de leur ADN. Comme l’ADN emballé dans une enveloppe de phage peut pénétrer dans les cellules de manière efficace des gènes sélectionnables sur les phages habituellement ne sont pas nécessaires.
Vecteurs plasmidiques :
La plupart des plasmides sont de petits cercles qui contiennent les éléments nécessaires pour la réplication de l’ADN, un ou deux gènes de résistance aux médicaments et une région d’ADN dans laquelle l’ADN étranger peut être inséré sans endommager les fonctions essentielles du plasmide.
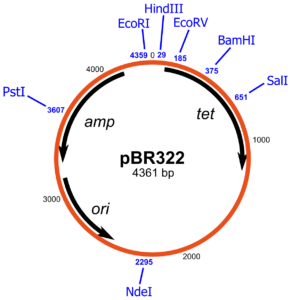
Un plasmide largement utilisé, pBR322, porte des gènes codant pour la résistance à la tetracycline et β-lactamase celle-ci confère une résistance à la pénicilline et aux analogues apparentés en clivant les médicaments dans le cycle lactame, ce qui les rend biologiquement inactifs. Des gènes conférant une résistance au chloramphénicol, à la tétracycline et kanamycine sont d’autres marqueurs de résistance aux médicaments couramment sélectionnables portés sur des plasmides.
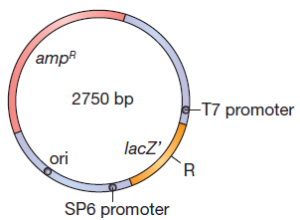
Un élément utile à avoir sur les plasmides est une origine de réplication de l’ADN à partir d’un phage simple brin. Quand une telle origine est activée par l’infection de tel phage, la cellule synthétise des quantités importantes d’un seul brin de l’ADN du plasmide. Ceci facilite le séquençage de l’ADN. Dans une expérience typique de clonage un plasmide est coupé dans une région non essentielle par une enzyme de restriction par exemple EcoRI et l’ADN monocaténaire étranger, coupé par EcoRI, est ajouté et les extrémités hybrides sont soudées ensemble. Seule une petite fraction des plasmides, soumis à ce traitement contiendra l’ADN inséré. La plupart auront recircularisé sans insertion d’ADN étranger. Comment les bactéries transformées c.à.d ceux dont les plasmides contiennent l’ADN inséré peuvent être distinguées des autres (ceux dont le plasmide reste sans ADN inséré)? Bien entendu dans certaines conditions une sélection génétique peut être utilisée pour activer uniquement la croissance des éléments transformés avec le fragment souhaité d’ADN inséré. Le plus souvent cela n’est pas possible et il devient nécessaire d’identifier des candidats qui contiennent l’ADN inséré.Une méthode pour identifier des candidats repose sur l’inactivation par l’ insertion d’un gène de résistance aux médicaments.
Par exemple, dans la résistance à l’ampicilline, le gène de la résistance sur pBR322 possède un seul site de clivage du plasmide par l’enzyme de restriction PstI. Heureusement le clivage par PstI génère des extrémités collantes et l’ADN peuvent être facilement soudées à ce niveau, après quoi il désactive le gène de la résistance à l’ampicilline. Le gène de résistance à la tétracycline sur le plasmide demeure intact et peut être utilisé pour la sélection des cellules transformées par le plasmide recombinant. Les colonies résultantes peuvent être testées en repérant sur une paire de plaques, l’une contenant de l’ampicilline, et l’autre sans ampicilline. Seul les mutants ampicilline sensibles, tétracycline-résistants vont contenir l’ADN étranger dans le plasmide alors que les ampicilline résistantess possèdent des plasmides re-circularisés sans insertion de l’ADN étranger.
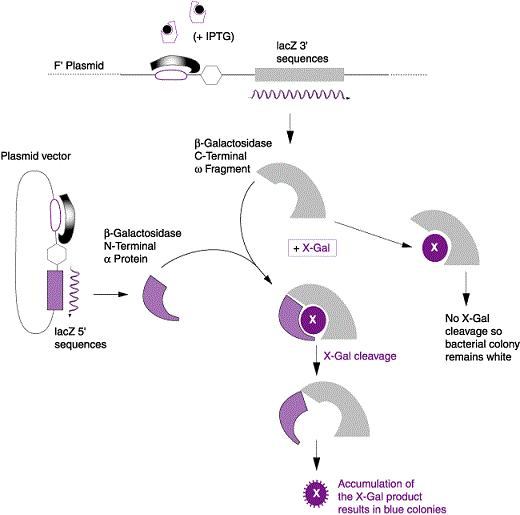
Une autre façon pour vérifier l’insertion d’ADN étranger, utilise le gène de ß-galactosidase. L’insertion d’ADN étranger dans le gène inactive l’enzyme qui peut être détectée en mettant les cellules transformées sur un milieu qui sélectionne la présence du plasmide et contient également des substrats de β-galactosidase qui produisent des colorants lorsqu’il hydrolyse ces substrats.
Un plasmide serait lourd s’il contenait la totalité des 3000 paires de bases du gène β galactosidase. Par conséquent seule une extrémité N-terminale du gène est placé sur le plasmide. Le reste de l’enzyme est codée par un segment inséré dans le chromosome des cellules hôtes. Les deux parties du gène synthétisent des domaines qui se lient entre eux pour donner l’enzyme actif. Ce phénomène inhabituel est appelé alpha-complémentation.
Des vecteurs de clonage sont conçus pour l’insertion d’ ADN étranger dans la partie N-terminale de la β-galactosidase. Une technique simple peut réduire considérablement la re-circularisation des molécules du vecteur sans insertion de l’ADN étranger. Si l’ADN du vecteur est traité par une phosphatase après coupure par l’enzyme de restriction, la re-circularisation devient impossible parce que l’extrémité 5′-PO4 requise pour l’ ADN ligase est absente. L’ADN étranger, cependant, contient une extrémité 5′-PO4 et par conséquent deux des quatre fragments d’ADN encadrant un fragment d’ADN étranger peuvent être soudés. Cet ADN est actif dans la transformation parce que les cellules réparent le pseudo restant à chaque extrémité du fragment inséré. Les vecteurs de clonage plasmides ou phages peuvent contenir un court tronçon de l’ADN contenant des sites de coupure unique pour plusieurs enzymes de restriction. Ces régions à liaisons multiples (polylinker) permettent le clivage par deux enzymes de sorte que les extrémités collantes résultantes ne sont pas auto-complémentaire. Donc le vecteur ne peut pas se re-circularisé et être resoudé sur lui-même mais quand un fragment d’ADN contenant les extrémités nécessaires et complémentaires aux extrémités du plasmide existe alors il peut le faire. Une génie génétique efficace nécessite que l’ADN plasmidique soit obtenu en grandes quantités. Certains plasmides maintiennent seulement trois ou quatre copies par cellule, tandis que d’autres plasmides ont 25 à 50 copies cellulaires. La plupart des plasmides ayant un nombre importants de copies peuvent être amplifiée du fait que le plasmide continue de se répliquer après que la synthèse protéique et la synthèse de l’ADN cellulaire ont cessé en raison d’une densité cellulaire élevée ou la présence d’inhibiteurs de la synthèse des protéines. Après amplification, une cellule contenant un tel plasmide peut contenir jusqu’à 3000 copies de plasmide.

La nature n’a pas livré des vecteurs plasmidiques prêt pour le génie génétique.Les vecteurs plasmidiques les plus utiles ont été eux-mêmes construits par génie génétique. Les matières de départ sont les plasmides R, ce sont des plasmides ou des éléments d’ADN à réplication autonome qui portent un ou plusieurs gènes de résistance aux médicaments. Les plasmides R sont la cause de graves problèmes médicaux, car diverses bactéries peuvent acquérir des plasmides R et ainsi devenir résistant aux médicaments normalement utilisés pour le traitement des infections . La conversion d’un plasmide R en un vecteur utile nécessite l’élimination de l’ADN étranger et l’élimination des multiples sites de clivage par des enzymes de restriction. Afin que l’ADN étranger puisse être cloné dans le plasmide, le plasmide doit posséder un seul site de clivage pour au moins une enzyme de restriction, et cela devrait être dans une région non essentielle. Dans la construction des vecteurs de clonage, des plasmides ont été digérés avec diverses enzymes de restriction. Le mélange résultant de fragments d’ADN a été hybridés ensemble par l’intermédiaire des extrémités auto-complémentaires, puis collées pour produire de nombreuses combinaisons de fragments possibles . Ces ADN ont été transférés dans des cellules. Seuls les nouveaux plasmides contenant au moins les segments d’ADN nécessaires à la réplication et à la résistance aux médicaments ont survécu et ont donné des colonies. Les plasmides souhaitables contenant uniquement les sites de clivage uniques pour des enzymes de restriction peuvent être identifiés par l’amplification et la purification de l’ADN suivie par des digestions test avec des enzymes de restriction suivi d’électrophorèse pour caractériser les Produits de la digestion. Le plasmide pBR322 possède une seule site de clivage pour plus de 20 enzymes de restriction. Parmi les plus communément utilisés sont Bam HI, Eco RI, Hind III, PstI, PvuII et Sali.
Un vecteur phagique pour les bactéries :
des vecteurs de phage et des vecteurs dérivés de phages sont utiles pour trois raisons:
-les phages peuvent transporter de plus grands fragments d’ADN insérés que des plasmides. Donc beaucoup moins de candidats transformés doivent être examinés pour trouver un clone désiré.
-L’efficacité d’infecter les cellules par l’ADN des phages reconditionnés est considérablement plus grande que l’efficacité de la transformation avec L’ADN de plasmide dans les cellules. Ceci est un facteur important quand un clone rare est recherché.
-Enfin, le phage lambda permet un procédé commode pour le criblage pour détecter le clone porteur du gène désiré. Cependant une fois qu’un segment désiré d’ADN a été cloné dans un phage la commodité de manipuler des plasmides, en partie en raison de leur petite taille, dicte que le segment soit sous-cloné dans un plasmide.
Vecteur de clonage basé sur le bactériophage M13 :
L’exigence la plus essentielle pour tout vecteur de clonage est qu’il ait un moyen de se répliquer dans la cellule hôte. Pour les vecteurs plasmidiques, cette exigence est facile à satisfaire, car des séquences d’ADN relativement courtes sont capables d’agir comme origines de réplication plasmidiques, et la plupart, sinon la totalité, des enzymes nécessaires à la réplication sont fournies par la cellule hôte. Des manipulations élaborées, telles que celles qui ont abouti à pBR322, sont donc possibles tant que la construction finale a une origine de réplication fonctionnelle et intacte. Avec des bactériophages tels que M13 et e, la situation en matière de réplication est plus complexe. Les molécules d’ADN phagique portent généralement plusieurs gènes essentiels à la réplication, y compris des gènes codant pour des composants de la couche protéique du phage et des enzymes réplicatives spécifiques de l’ADN phagique. La modification ou la suppression de l’un de ces gènes altèrent ou détruisent la capacité de réplication de la molécule résultante. Il y a donc beaucoup moins de liberté pour modifier les molécules d’ADN phagique, et généralement les vecteurs de clonage de phages ne sont que légèrement différents de la molécule parente.
Les problèmes de construction d’un vecteur de phages sont illustrés en considérant M13. Le génome M13 normal a une longueur de 6,4 kb, mais la plus grande partie de ce génome est occupée par dix gènes étroitement liés, chacun étant essentiel à la réplication du phage. Il n’y a qu’une seule séquence intergénique de 507 nucléotides dans laquelle un nouvel ADN pourrait être inséré sans perturber l’un de ces gènes, et cette région comprend l’origine de réplication qui doit elle-même rester intacte. Clairement, il n’y a qu’une possibilité limitée de modifier le génome M13.
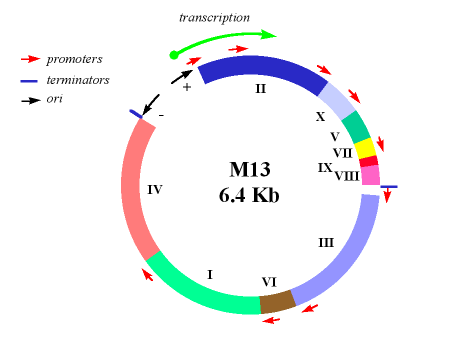
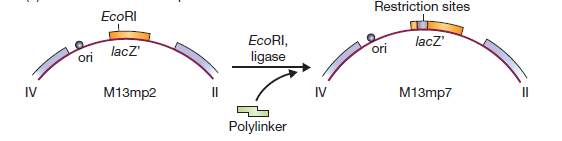
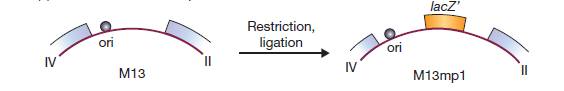
La première étape de construction d’un vecteur M13 consistait à introduire le gène lacZ ‘dans la séquence intergénique. Cela a donné M13mp1, qui forme des plaques bleues sur de l’agar X-gal.
M13mp1 ne possède pas de sites de restriction uniques dans le gène lacZ ‘. Cependant, il contient l’hexanucléotide GGATTC près du début du gène. Un seul changement de nucléotide rendrait ce GAATTC, qui est un site EcoRI.
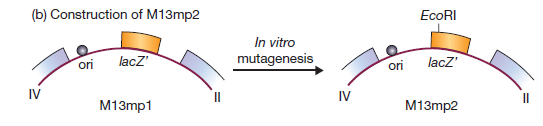
Cette altération a été réalisée en utilisant la mutagenèse in vitro, résultant en M13mp2
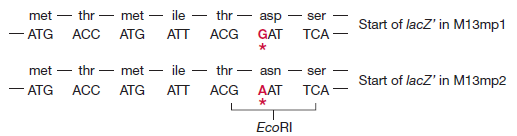
M13mp2 a un gène lacZ′ légèrement modifié (le sixième codon spécifie maintenant l’asparagine à la place de l’acide aspartique), mais l’enzyme β-galactosidase produite par les cellules infectées par M13mp2 est encore parfaitement fonctionnelle. L’étape suivante dans le développement des vecteurs M13 consistait à introduire des sites de restriction supplémentaires dans le gène lacZ’. Ceci a été réalisé en synthétisant dans le tube à essai un oligonucléotide court, appelé polylinker, qui consiste en une série de sites de restriction et a des extrémités collantes EcoRI.
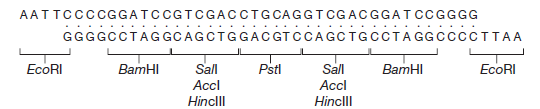
Ce linker multisite a été inséré dans le site EcoRI de M13mp2, pour donner M13mp7, un vecteur plus complexe avec quatre sites de clonage possibles (EcoRI, BamHI, Sali et PstI). Le polylinker est conçu pour ne pas perturber totalement le gène lacZ': un cadre de lecture est maintenu dans tout le polylinker, et une enzyme β-galactosidase fonctionnelle bien que modifiée est encore produite. Les vecteurs M13 les plus sophistiqués ont des polylinkers plus complexes insérés dans le gène lacZ’. Un exemple est M13mp8, qui a la même série de sites de restriction que le plasmide pUC8 . Comme avec le vecteur plasmidique, un avantage de M13mp8 est sa capacité à prendre des fragments d’ADN avec deux extrémités cohésives différentes.