Chapter 1 : recombinant DNA technology
Introduction
To facilitate the study of genes, they must be isolated and amplified. One method of isolation and amplification of a gene of interest is to clone the gene by inserting it into another DNA molecule that serves as a vehicle or vector that can be replicated in living cells. When these two DNAs of different origin are combined, the result is a recombinant DNA molecule. The recombinant DNA molecule is placed in a host cell, either prokaryotic or eukaryotic. The host cell then replicates (producing a clone), and the vector with its foreign piece of DNA also replicates. The foreign DNA thus becomes amplified in number, and following its amplification can be purified for further analysis.
In 1962, Allan Campbell noted that the linear genome of bacteriophage λ forms a circle upon entering the host bacterial cell, and a recombination (breaking and rejoining) event inserts the phage DNA into the host chromosome. Reversal of the recombination event leads to normal excision of the phage DNA. Further analysis revealed that phage λ had short regions of single-stranded DNA whose base sequences were complementary to each other at each end of its linear genome. These single-stranded regions were called “cohesive” (cos) sites. Complementary base pairing of the cos sites allowed the linear genome to become a circle within the host bacterium. The idea of joining DNA segments by “cohesive sites” became the guiding principle for the development of genetic engineering. With the molecular characterization of restriction and modification systems in bacteria, it soon became apparent that the ideal engineering tools for making cohesive sites on specific DNA pieces were already available in the form of restriction endonucleases.
Early on, Salvador Luria and other phage workers were intrigued by a phenomenon termed “restriction and modification.” Phages grown in one bacterial host often failed to grow in different bacterial strains (“restriction”). However, some rare progeny phages were able to escape this restriction. Once produced in the restrictive host they had become “modified” in some way so that they now grew normally in this host. The entire cycle could be repeated, indicating that the modification was not an irreversible change. For example, phage λ grown on the C strain of Escherichia coli (λ·C) were restricted in the K-12 strain (the standard strain for most molecular work). However, the rare phage λ that managed to grow in the K-12 strain now had “K” modification (λ·K). These phages grew normally on both C and K-12; however, after growth on C, the phage λ with “C” modification (λ·C) was again restricted in K-12. Thus, the K-12 strain was able to mark its own resident DNA for preservation, but could eliminate invading DNA from another distantly related strain. In 1962, the molecular basis of restriction and modification was defined by Werner Arber and co-workers.
Restriction system :
After demonstrating that phage λ DNA was degraded in a restricting host bacterium, Arber and co-workers hypothesized that the restrictive agent was a nuclease with the ability to distinguish whether DNA was resident or foreign. Six years later, such a nuclease was biochemically characterized in E. coli K-12 by Matt.
Meselson and Bob Yuan. The purified enzyme cleaved λ·C-modified DNA into about five pieces but did not attack λ·K-modified DNA. Restriction endonucleases (also referred to simply as restriction enzymes) thus received their name because they restrict or prevent viral infection by degrading the invading nucleic acid.
Modification system
At the time, it was known that methyl groups were added to bacterial DNA at a limited number of sites. Most importantly, the location of methyl groups varied among bacterial species. Arber and colleagues were able to demonstrate that modification consisted of the addition of methyl groups to protect those sites in DNA sensitive to attack by a restriction endonuclease. In E. coli, adenine methylation (6-methyl adenine) is more common than cytosine methylation (5-methyl cytosine)
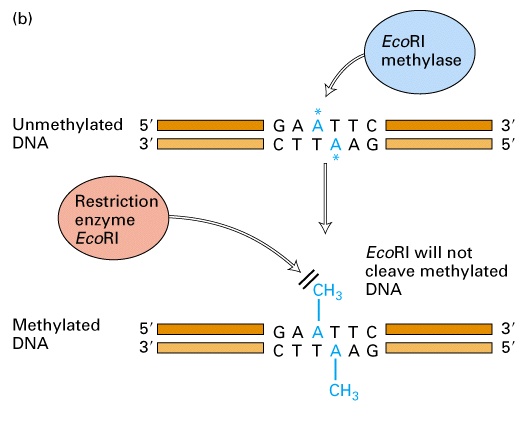 Methyl-modified target sites are no longer recognized by restriction endonucleases and the DNA is no longer degraded. Once established, methylation patterns are maintained during replication. When resident DNA replicates, the old strand remains methylated and the new strand is unmethylated. In this hemimethylated state, the new strand is quickly methylated by specific methylases. In contrast, foreign DNA that is unmethylated or has a different pattern of methylation than the host cell DNA is degraded by restriction endonucleases.
Methyl-modified target sites are no longer recognized by restriction endonucleases and the DNA is no longer degraded. Once established, methylation patterns are maintained during replication. When resident DNA replicates, the old strand remains methylated and the new strand is unmethylated. In this hemimethylated state, the new strand is quickly methylated by specific methylases. In contrast, foreign DNA that is unmethylated or has a different pattern of methylation than the host cell DNA is degraded by restriction endonucleases.
The first cloning experiments
Hamilton Smith and co-workers demonstrated unequivocally that restriction endonucleases cleave a specific DNA sequence. Later, Daniel Nathans used restriction endonucleases to map the simian virus 40 (SV40) genome and to locate the origin of replication. These major breakthroughs underscored the great potential of restriction endonucleases for DNA work. Building on their discoveries, the cloning experiments of Herbert Boyer, Stanley Cohen, Paul Berg, and their colleagues in the early 1970s ushered in the era of recombinant DNA technology. One of the first recombinant DNA molecules to be engineered was a hybrid of phage λ and the SV40 mammalian DNA virus genome. In 1974 the first eukaryotic gene was cloned. Amplified ribosomal RNA (rRNA) genes or “ribosomal DNA” (rDNA) from the South African clawed frog Xenopus laevis were digested with a restriction endonuclease and linked to a bacterial plasmid. Amplified rDNA was used as the source of eukaryotic DNA since it was well characterized at the time and could be isolated in quantity by CsCl-gradient centrifugation. Within oocytes of the frog, rDNA is selectively amplified by a rolling circle mechanism from an extrachromosomal nucleolar circle . The number of rRNA genes in the oocyte is about 100- to 1000-fold greater than within somatic cells of the same organism. To the great excitement of the scientific community, the cloned frog genes were actively transcribed into rRNA in E. coli. This showed that recombinant plasmids containing both eukaryotic and prokaryotic DNA replicate stably in E. coli. Thus, genetic engineering could produce new combinations of genes that had never appeared in the natural environment, a feat which led to widespread concern about the safety of recombinant DNA work
Cutting and joining DNA
Two major categories of enzymes are important tools in the isolation of DNA and the preparation of recombinant DNA: restriction endonucleases and DNA ligases. Restriction endonucleases recognize a specific, short, nucleotide sequence on a double-stranded DNA molecule, called a restriction site, and cleave the DNA at this recognition site, depending on the type of enzyme. DNA ligase joins two pieces of DNA by forming phosphodiester bonds.
Major classes of restriction endonucleases
There are three major classes of restriction endonucleases. Their grouping is based on the types of sequences recognized, the nature of the cut made in the DNA, and the enzyme structure:
– Type I and III restriction endonucleases are not generally used for gene cloning because they cleave DNA at sites other than the recognition sites and thus cause random cleavage patterns.
– type II endonucleases are widely used for mapping and reconstructing DNA in vitro because they recognize specific sites and cleave just at these sites . In addition, the type II endonuclease and methylase activities are usually separate, single subunit enzymes. Although the two enzymes recognize the same target sequence, they can be purified separately from each other
Nomenclature :
Restriction endonucleases are named for the organism in which they were discovered, using a system of letters and numbers. For example, HindIII was discovered in Haemophilus influenza strain d and III is for the third enzayme of that type. Sma I is from serratia marcescens , Eco RI is Escherichia coli( strain R) and BamHI is from Bacillus amyloliquefaciens ( strain H ). Over 3000 type II esrtriction endonucleases have been isolated and characterised to date.
Recognition sequences for type II restriction endonucleases :
Each orthodox type II restriction endonuclease is composed of two identical polypeptide subunits that join together to form a homodimer. These homodimers recognize short symmetric DNA sequences of 4–8 bp. Six base pair cutters are the most commonly used in molecular biology research. Usually, the sequence read in the 5′ → 3′ direction on one strand is the same as the sequence read in the 5′ → 3′ direction on the complementary strand. Sequences that read the same in both directions are called palindromes . Figure 8.3 shows some common restriction endonucleases and their recognition sequences. Some enzymes, such as EcoR1, generate a staggered cut, in which the single-stranded complementary tails are called “sticky” or cohesive ends because they can hydrogen bond to the singlestranded complementary tails of other DNA fragments. If DNA molecules from different sources share the same palindromic recognition sites, both will contain complementary sticky ends (single-stranded tails) when digested with the same restriction endonuclease. Other type II enzymes, such as SmaI, cut both strands of the DNA at the same position and generate blunt ends with no unpaired nucleotides when they cleave the DNA.
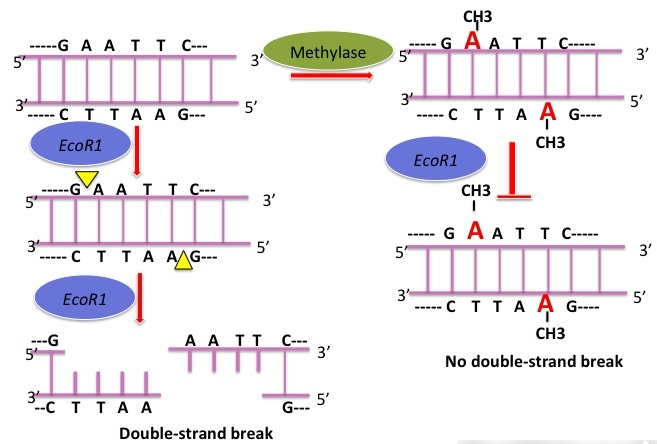
Restriction endonucleases exhibit a much greater degree of sequence specificity in the enzymatic reaction than is exhibited in the binding of regulatory proteins, such as the Lac repressor to DNA . For example, a single base pair change in a critical operator sequence usually reduces the affinity of the Lac repressor by 10- to 100-fold, whereas a single base pair change in the recognition site of a restriction endonuclease essentially eliminates all enzymatic activity.
Like other DNA-binding proteins, the first contact of a restriction endonuclease with DNA is nonspecific. Nonspecific binding usually does not involve interactions with the bases but only with the DNA sugar–phosphate backbone. The restriction endonuclease is loosely bound and its catalytic center is kept at a safe distance from the phosphodiester backbone. Nonspecific binding is a prerequisite for efficient target site location. For example, BamHI moves along the DNA in a linear fashion by a process called “sliding.” Sliding involves helical movement due to tracking along a groove of the DNA over short distances (< 30–50 bp). This reduces the volume of space through which the protein needs to search to one dimension. However, the “random walk” nature of linear diffusion gives equal probabilities for forward and reverse steps, so if the distances between the nonspecific binding site and the recognition site are large (> 30–50 bp), the protein
would return repeatedly to its start point. The main mode of translocation over long distances is thus by “hopping” or “jumping.” In this process, the protein moves between binding sites through three-dimensional space, by dissociating from its initial site before reassociating elsewhere in the same DNA chain. Because of relatively small diffusion constants of proteins, most rebinding events will be short range “hops” back to or near the initial binding site. In the example of BamHI, once the target restriction site is located, the recognition process triggers large conformational changes of the enzyme and the DNA (called coupling), which leads to the activation of the catalytic center. In addition to indirect interaction with the DNA backbone, specific binding is characterized by direct interaction of the enzyme with the nitrogenous bases.
All structures of orthodox type II restriction endonucleases characterized by X-ray crystallography so far show a common structural core composed of four conserved β-strands and one α-helix . In the presence of the essential cofactor Mg2+, the enzyme cleaves the DNA on both strands at the same time within or in close proximity to the recognition sequence (restriction site). The enzyme cuts the duplex by breaking the covalent, phosphodiester bond between the phosphate of one nucleotide and the sugar of an adjacent nucleotide, to give free 5′-phosphate and 3′-OH ends. Type II restriction endonucleases do not require ATP hydrolysis for their nucleolytic activity. Although there are a number of models for how this nucleophilic attack on the phosphodiester bond occurs, the exact mechanism by which restriction endonucleases achieve DNA cleavage has not yet been proven experimentally for any type II restriction endonuclease.
DNA ligase :
The study of DNA replication and repair processes led to the discovery of the DNA-joining enzyme called DNA ligase. DNA ligases catalyze formation of a phosphodiester bond between the 5′-phosphate of a nucleotide on one fragment of DNA and the 3′-hydroxyl of another . This joining of linear DNA fragments together with covalent bonds is called ligation. Unlike the type II restriction endonucleases, DNA ligase requires ATP as a cofactor.
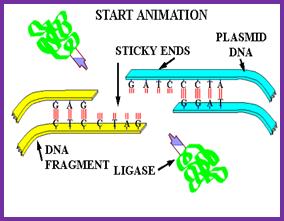
Because it can join two pieces of DNA, DNA ligase became a key enzyme in genetic engineering. If restriction-digested fragments of DNA are placed together under appropriate conditions, the DNA fragments from two sources can anneal to form recombinant molecules by hydrogen bonding between the complementary base pairs of the sticky ends. However, the two strands are not covalently bonded by phosphodiester bonds. DNA ligase is required to seal the gaps, covalently bonding the two strands and regenerating a circular molecule. The DNA ligase most widely used in the lab is derived from the bacteriophage T4. T4 DNA ligase will also ligate fragments with blunt ends, but the reaction is less efficient and higher concentrations of the enzyme are usually required in vitro. To increase the efficiency of the reaction, researchers often use the enyzme terminal deoxynucleotidyl transferase to modify the blunt ends. For example, if a single-stranded poly(dA) tail is added to DNA fragments from one source, and a singlestranded poly(dT) tail is added to DNA from another source, the complementary tails can hydrogen bond. Recombinant DNA molecules can then be created by ligation.