GENETIC ENGINEERING
Definition:
Direct and deliberate modification of an organism’s genome.
Spotlights and topics in genetic engineering
Hybridization
Genetic tools
DNA analysis (electrophoresis and PCR)
Recombinant DNA technology
Gene therapy
Genome analysis (fingerprinting and microarray)
HYBRIDIZATION
Allows identification of an unknown organism by looking at its DNA sequence. Hybridization use a DNA that binds complementary to the DNA of only one specific species according to base pair rules. If it Binds to the DNA of an unknown organism we can identify the unknown one.
GENETIC TOOLS
Enzymes for dicing, splicing, reversing nucleic acids,allows the analysis of DNA. Enzymes that can be used as tools:
1) Helicase
2) Ligase
3) Polymerase
4) Reverse transcriptase
5) Restriction endonucleases
Helicase:
Helicase, unwinds double stranded DNA and we can do it in test tube. The other possibility is heating (also denature or unwinds the double stranded DNA).
Ligase:
seals gaps in DNA, so it seals two pieces of DNA together like a molecular glue.
Polymerase:
reads one strand of DNA and synthesize a complementary anti parallel strand. RNA polymerases reads DNA and makes complementary RNA.
Reverse transcriptase:
Reverse transcriptase makes a DNA copy from RNA. That’s a very unusual property and ability and the reason why it’s called reverse transcriptase is that it’s doing an opposite thing. Because transcription makes an RNA from DNA. In fact, this enzyme is not found in any cells, it is only found in a couple of viruses. Only a one small family of viruses, carries this enzyme, HIV is one of these viruses.
Restrictions endonucleases:
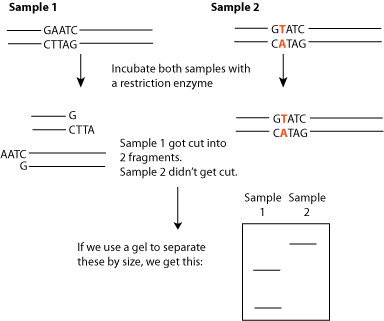
Recognizes specific DNA sequences and cut at that sequence. And they are extremely specific. There are a whole branch of different restriction endonucleases and each one cuts at a very specific DNA sequence (they are a molecular scissors for DNA). They are very specific. There are hundreds of restriction endonucleases but each one cut a specific site.
Imagine a sequence of DNA ,And we cut this with three different restriction endonucleases. We will have fragments.


If we have 2 enzymes with 2 cut sites, we will have three fragments.
After these scissions we can put them on gel electrophoresis,a method for separating DNA fragments according to their size:
How it works?
We take a gel and we put our samples at the top of gel and then we expose it to a charge (normally DNA has a negative charge because it has many phosphate groups)Therefor DNA begins to move towards the positive charge.How fast does it move depends on the size of the fragments?
Large fragments travel slowly, small fragments travel quickly, so it separates DNA fragments according to their size .
We will see an example ,checking a DNA sequence.We have a piece of DNA and we want to check the sequence…
5’TTC AAGCTTACGAAGGATTCGTATAC 3′
What we can do is we cut by restriction endonucleases and we know that enzyme 1 cuts at AAGC.
And the enzyme 2 cuts at AGGA.So if we add these two different restriction endonucleases to this DNA we can predict how many fragments we expect to find:
We must find three fragments after the procedure (after complete digestion) but we also can know the size of each of those fragments.We know that this first fragment should be only three nucleotides long and the second fragment should be a five nucleotides long and the third fragment must be nine nucleotides long. Now how we can see the size of fragments?
We can run it on gel electrophoresis by putting our sample on the top of the gel and adding in, a standard mix, side by side .
How fast, each piece of DNA, moves depends on its size, smaller fragments move faster, larger fragments move more slowly.

3, 5, 9 nucleotides are the sizes that we can expect. So restriction endonucleases can be used as a way of checking a sequence.
Polymerase chain reaction (PCR) amplifies DNA:
PCR amplifies DNA: it is like a copy machine for DNA . The DNA polymerase chain reaction copies DNA and this is important because,we may have a DNA sample but we don’t have enough to analyze.For example we’re going to do gel electrophoresis,we will need a considerable amount of DNA to run those different tests.
How it works?
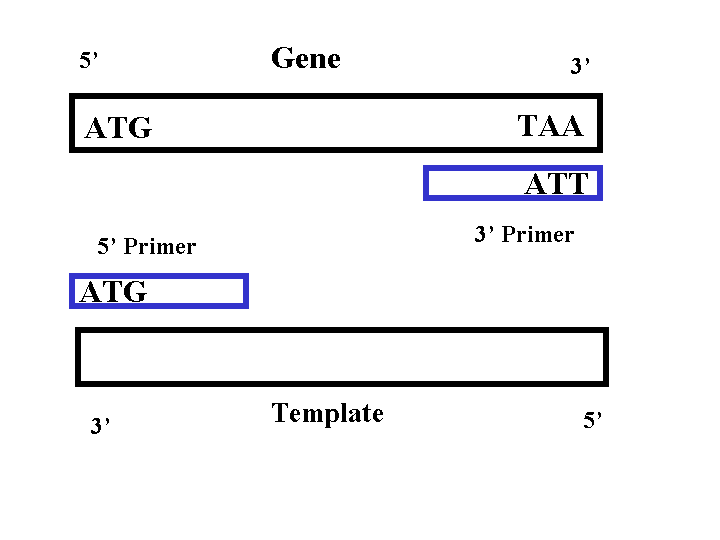
We add template DNA, DNA polymerase III, primers, nucleotide mix in a PCR tube and then we add our PCR tube to a PCR machine,and the PCR machine changes the temperature in order to copy DNA.

Here we have our template, PCR machine heat up to 94° (which is high enough to denature the DNA and the two strands separate).Then PCR machine switches to 55° (at which temperature primers will attach),Then the machine switches to 72° (at this temperature DNA polymerase attaches the primers and begins to copy them). by the end we will have 2 copies.
Then the machine goes back to 94° (denaturation) and 55° ( primer attachment) and 72° ( copy extension ), and the repetition over and over so PCR, copy and increase the DNA synthesis by changing temperature.


Before PCR machine we were doing that by incubators (one at 94°, one at 55° and one 72°)
Recap of DNA analysis methods
1) Enzymes can be used as molecular tools.
2) Gel electrophoresis : separates DNA fragments based on size.
3) Polymerase chain reaction: Amplifies DNA (make copies of the DNA) and Good primer design allows the isolation and amplification of a particular gene out of the entire genome
Using PCR to isolate and amplify a gene from the genome
Primers are designed to bind a specific place in the DNA according to complementary base-pair rules .Since DNA polymerase III, can only start DNA replication, where the primer is attached, the primer attachment site controls what part of DNA, is replicated by the PCR. By designing primers to bind by complementarity on each side of the gene of interest we can specify the PCR to isolate and amplify just that gene and nothing else
Recombinant DNA technology
Transferring DNA from one organism to another, in this process, there are a genetic donor , a vector , a host.The vector, transfers the gene, from the donor to the host. There are different vectors, the most common vector used, is the plasmid (a small circular piece of DNA),the other possibility would be a virus, they can also transfer DNA from one organism to another.
An example of a cloning vector is pBR 322: This is a plasmid that is used currently
There are many plasmids but they all have some common factors :
1) All vectors must have an (ORI) which is a specific DNA sequence of 50 – 100 base pairs that must be present in a plasmid for it to replicate. Host-cell enzymes bind to ORI, initiating replication of the circular plasmid. Once DNA replication is initiated at ORI, it continues around the circular plasmid regardless of its nucleotide sequence . Thus any DNA sequence inserted into such a plasmid is replicated along with the rest of the plasmid DNA.
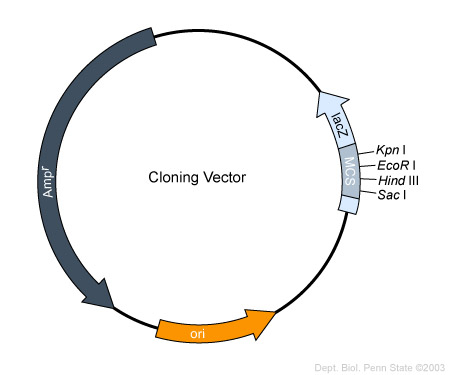
2)Plasmid also must have some sort of Promoter: where the gene can be transcribed.
3)Plasmid must also have a room for gene insertion. In other words it can’t be too large and it also should have a whole bunch at different place where we could cut the plasmid and insert our gene of interest.
4)Reporter gene : we must also have a reporter gene which inform us whether or not the gene is there.
Very common reporter gene would be antibiotic resistance genes so PTR322 carriers tetracycline resistance gene as well as an ampicillin resistance gene
In other words normally E-coli is killed by ampicillin or tetracyclin but if that E coli has PBR 322 inside of it it’s going to survive an ampicillin or tetracycllin.
Example:
If we want to move a gene for human insulin into E coli:the first step is to isolate an amplify your gene of interest.How are we going to isolate and amplify our gene of interest (which is the human insulin gene)?
We will do it by PCR. By designing correctly our primers, we can copy only the human insulin gene.
we take a plasmid like pTR 322, and we put your gene of interest (Human Insulin Gene) into the PBR 322 .We cut a hole in the plasmid by restriction endonuclease,then we seal the insulin gene in the plasmid by ligase .
So, PCR ⇒restriction endonuclease ⇒ligase
Now, we have our plasmid with our gene of interest on it.Then, we put our cloning host E coli and our vector pTR 322 in the test tube and we will do the transformation.
Remember, that transformation is not a common event (it is not a very efficient process).What can we do to increase the efficiency of transformation?
What can we do? to increase that bacteria taking in that plasmid? We know that some chemical treatments and the electricity encourage the bacteria to take in the plasmid.
Remember that if we put all the bacteria in normal culture medium all bacteria will grow But if we put them in the medium with ampicillin or tetracycline, only the E coli’s having the plasmid will develope.
Biochemical products of recombinant DNA technology
Enables the large scale manufacturing of life-saving hormones, enzymes and vaccines:
Insulin for diabetes
Human growth hormone for dwarfism
Erythropoietin for anemia
Factor 8 for hemophilia
HBV vaccine
Genetically modified organisms
1) Recombinant microbes:
Microbes synthesizing a wide variety of proteins used in medicine, agriculture, industry, etc.
2) Transgenic plants:
Plants that synthesize vitamins, medicines or agriculturally important substances
Agrobacterium is interesting because it naturally transforms plant cells. (it is part of it’s normal natural behavior).The particular plasmid that agrobacterium contains is Ti plasmid.
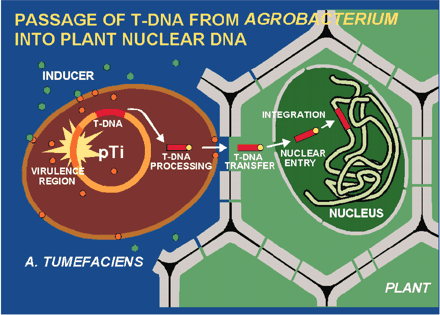
If an agrobacterium encounters a plant it pushes his plasmid into the plant cell, so it transforms the plant cell (this is a normal and natural behavior).Why the agrobacterium does that?
Because the Ti plasmid (tumor inducing plasmid), encodes all of the information, for making agro’s favorite food source. So, agrobacterium comes up to a plant cell, pushes his plasmid to the plant cell and the plant begins to produce the food that agro wants to eat. But, how it’s useful for us?
If we put our gene of interest on to the Ti plasmid and put the Ti plasmid into agro then agro will transmit that gene to the plant cells.
Let’s imagine that we will create a tomato plant that expresses human insulin, so our gene of interest is human insulin. All we have to do is isolate and amplify (by PCR) the human insulin gene from human genetic donor. By Good primer design we can copy only the human insulin gene. next we cut the TI plasmid by restriction endonucleases, and with the ligase we can seal the insulin gene on the plasmid.
All after is naturally done by agrobacterium.Agrobacterium easily infects all of the gross plants including potatoes, tomatoes, bananas………………
Transgenic animals:
Animals synthesizing medicines or providing models for human diseases
(cystic fibrosis is a disease of human and we can not study easily) ,so we must create a transgenic animal with the gene of cystic fibrosis.
Let’s see how we can create transgenic animals:
First, we collect embryos and culture them and then we inject our gene of interest in the embryo, and we hope that this DNA of this embryo will integrate the gene of interest into it’s genome. The success rate is not high.
And, then we inject this embryo to a female and then we will wait until a transgenic animal will born
What’s interesting?
1) We can create animal models for human diseases
2) We can create animals with humans genes for production of hormones
And probably we can grow human organs by this model.