Methods of separation
One job of a chemist is to analyse samples of chemicals. To determine the composition of the sample, a separation of the different elements is necessary to avoid interferences between the species.
It can be of importance to determine which chemical elements are present, in which quantities and oxidation state. Several methods of separation can fulfil the needs of the chemist to obtain a good separation. In most of the cases, the sample must be a solution the solvent of which does not contain any interfering species.
The separation methods are based on the affinity of species for different solvents, on equilibrium processes and on kinetics. Some methods allow to get the sample back and some destroy it.
Extraction
This method is based on the affinity of species with two different immiscible liquid phases. One phase is aqueous and the second one is organic. The experiment is performed in a separatory funnel (or sep funnel). It is a piece of glass or plastic shaped like a reversed cone with a valve on the bottom and an opening at its top. A cap can close the funnel to allow a vigorous mixing.

The sample is prepared in one of the two phases and is placed in the extraction funnel by its top opening. The second phase is added (the order of insertion of the two phases does not really matter). As the phases are immiscible, the exchanges of particles between the phases can only be made at the interface and charged species do not move into the organic phase: their solubility in any organic phase is very low. To improve the exchanges, the liquids are mixed by shaking the funnel and by turning it upside down. It increases the surface of contact between the two phases, thus the exchange is improved. During the mixing phase, it is recommended to open briefly the funnel (by the cap or by the valve) to release an eventual excess of pressure. Remember some security notions: it can be necessary to open the funnel under a hood and do not put your nose or an eye just above the opening when you release the pressure. Next, the system is put to rest until the interface is thin and clear. The phase of lowest density is at the top and the densest is at the bottom. Usually, the organic phase it above the aqueous phase because the density of organic solvents is usually lower than the density of the water (oil floats on water). We can thus remove the aqueous phase out of the funnel by opening the valve at the bottom of the flask.
Distribution of the species
At the equilibrium, each species is distributed between the two phases. We can define a coefficient of distribution KD that is the ratio between the concentration of a species in the organic phase and in the aqueous phase:

If we don’t specifically consider the equilibrium, it is the distribution ratio D.
![]()
For the concentrations of the species, we consider all the form of a given species. Reactions often depend on the solvent and may occur in one phase but not in the other one. For instance, an acid in the aqueous phase can be in its acidic form HA or in the conjugated base form A–. The solubility of ions in organic phases being very low, the exchange of species between the two phases is limited to HA.
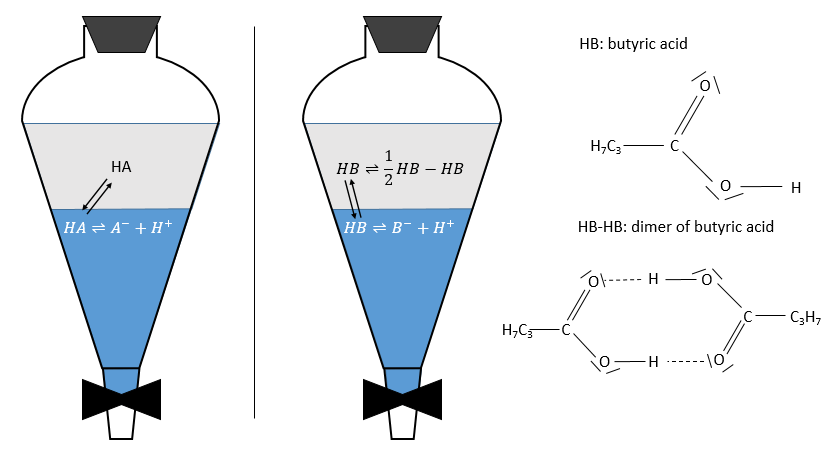
As a result, A– is not exchanged and remains in the aqueous phase while the neutral species HA has a concentration balanced between the aqueous phase and the organic phase. So, the distribution ratio of the acid is
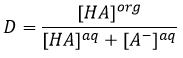
As a result, the distribution ratio may depend on the acidity of the aqueous phase. Equilibrium reaction can also specifically occur in the organic phase, as it is showed with the example of the butyric acid HB (see the right of the previous figure). In the organic phase, this carboxylic acid can form a dimer HB-HB, connecting two HB together by two hydrogen bonds. The dimer is stable in the organic phase but it is not the case in the aqueous phase where the acid can lose its proton. In this case, the distribution ratio will be
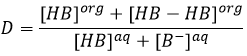
The choice of the solvents is thus crucial for a good extraction. The solvents can be polar or nonpolar depending on the separation that has to be done, but they must not be miscible.
Extraction of organic species
To extract an organic compound out of the organic phase, we want to get charged species. The neutral species are shared between the two phases but the ions remain in the aqueous phase. If the compound is not ionisable, we can only play on the nature of the solvent to expect the best extraction, i.e. when KD is small. If the compound can be dissociated, we can tune the acidity of the aqueous solution to optimise the extraction.
The acetic acid has a pKa of 4.7.
![]()
At this pH, in the aqueous solution, there will be as much CH3COOH as CH3COO–. The acetate can be extracted in the aqueous phase. If we use a stronger acid, a majority of the acetic acid that penetrates in the aqueous solution will dissociate to form acetate that can be extracted.
Another case is the case of amphoteric species. These species can play de role of a donor or an acceptor of proton. The oxine (8-Hydroxyquinoline) is an amphoteric.

At pH<5, the nitrogen is protonated, giving a positive charge to the compound that can now be extracted. At pH>10, the hydroxyl group loses its proton, leading to a negatively charged species that can also be extracted.
Between 5<pH<10, oxine remains in the organic phase.
Extraction of metals
The metallic cations can be extracted from aqueous solutions. In this case, we want to obtain a great value of D. To transfer the metals from the aqueous phase, their charge has to be hidden by the formation of strong ionic pairs or by the formation of coordination complexes. The oxine can form a complex with metals to stabilise them in the organic phase. For instance, if we want to extract aluminium from an aqueous solution, the oxine can travel from the organic phase to the aqueous phase. In good conditions of pH, the oxine loses its proton. In its basic form, the oxine forms a complex with the ions of aluminium, a complex that can now be transferred in the organic phase.
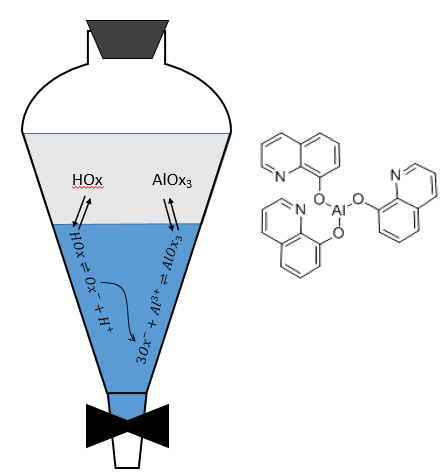
Fe3+ can form an ionic pair in a concentrated solution of HCl. The ionic pair (FeCl4)–H3O+ can move into the organic phase. Note that this product of the reaction moves out of the aqueous solution. The equilibrium is thus shifted to the right.
Another way to shift the equilibrium to the right is to increase the pH of the aqueous solution. It is possible to extract specifically several metals depending on the pH of the aqueous solution. The following figure represents the extraction of various metals by the formation of dithizomates in function of the pH.
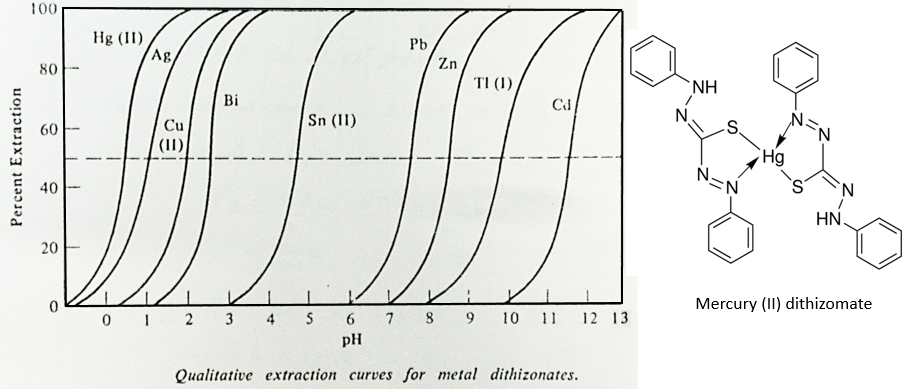
One can see that the extraction of Pb starts at pH>6 while Ag is already fully extracted at pH=3. The separation of Cu (II) from Ag will require another method of separation.
Extracted fraction
Let’s consider the extraction of the butyric acid. The distribution ratio D is the ratio of its concentration in the organic phase (ether) over its concentration in the aqueous phase (water). After the extraction, 3g of butyric acid is in the organic phase and 1g is still in the aqueous phase.
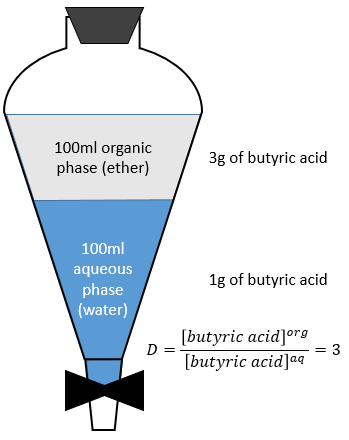
The extracted fraction F is the quantity of the acid in the organic phase over the total quantity of the acid:

F=3/4 if the volumes of the aqueous and organic phases are equal. The ratio of volumes RV is the volume of the organic phase over the one of the aqueous phase. If we consider a large volume of solvent with regard of the volume of the species to separate, the initial volumes of solvents put in presence can be used with a good approximation. Injecting RV in the expression of F, we obtain the following expression:
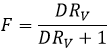
We can conclude that an extraction has better results when D or RV is large.
The remaining fraction G is the fraction of the sample that has not been transferred to the organic phase and is simply
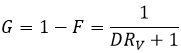
Now if two compounds A and B are to be separated, we have to design the experiment such as the multiplication of DA by DB is equal to 1. Indeed, if DA and DB are large but DA>>>DB, the separation will not be efficient. Image that DA=1000 and DB=20. A has a better extraction than B, but both species are extracted: the extracted fractions are, considering identical volumes of phases
![]()
Despite the huge difference of D, the separation is bad. Now, if DA=10 and DB=0.1, the extraction of both compounds is lower than in the previous example but the separation is way better:
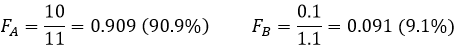
This time, 91% of A are in the organic phase for only 9% of B.
To obtain a better separation, we can repeat the extraction starting with the organic phase we just extracted. Let’s come back to our first example of the butyric acid. For this compound, we had found D=3. We can play on the volumes of the phase to modify the efficiency of the extraction. If we put half the volume of ether, then RV=0.5. The extracted fraction will be
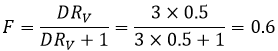
Out of the 4 initial grams, 1.6g are in the aqueous phase and 2.4g are in the organic phase. The extraction was batter with a larger volume of ether. However, if we do a second extraction with the same conditions, F is again 0.6 and we will have 0.64g remaining in the aqueous phase.
We have done two extractions with 50ml of ether instead of one extraction with 100ml of ether. So with the same volume, but with two extractions, we extracted more butyric acid (3.36g vs 3g). Several extractions with a small volume are thus more efficient than one extraction with a large volume. After n extraction, the remaining fraction is

Craig Apparatus
This method has been optimized into the apparatus of Craig. Dozens to hundreds of H shaped tubes are connected. The next figure shows one tube.
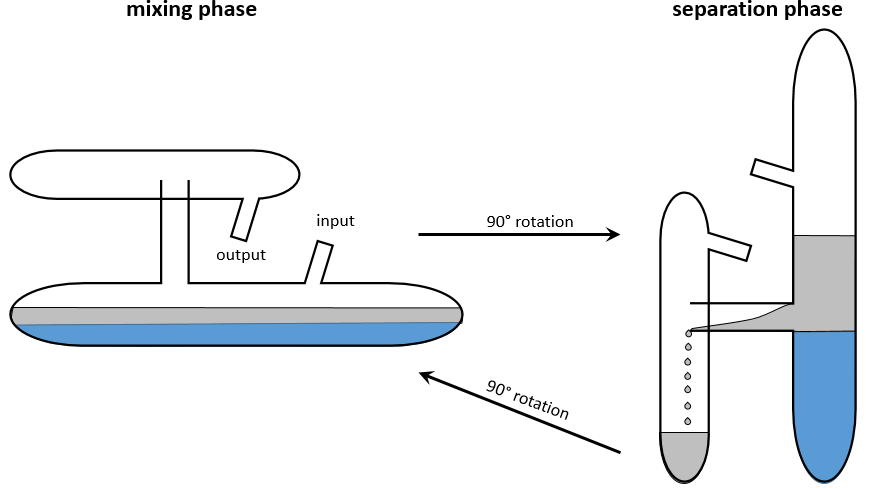
Mechanical arms mix the small tubes during the mixing phase and tilt them to transfer the top phase in the small compartment of the H shaped tube. Once the transfer is done, the tubes are put back in their initial position. This rotation causes the emptying of the small compartment in the next tube. The process is repeated in all the tubes simultaneously.
Initially, the sample is in the first tube (noted 0) and the bottom phase is already in all the tubes.
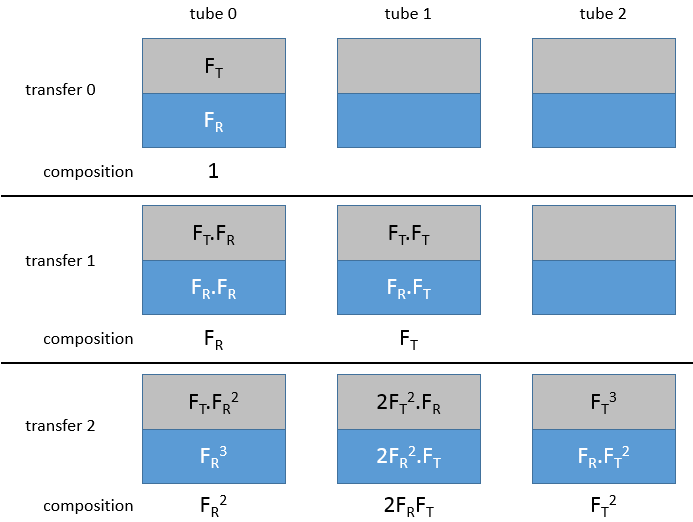
Initially, all the substrate is in the tube 0, half of it in the organic phase and the other half in the aqueous phase. The organic phase is transferred into the tube 1. We can define a remaining fraction FR, that is the fraction of the sample that remains in the tube during a transfer, and a transferred fraction FT that is the fraction that is transferred in the next tube.. After the first transfer, there is FR in t0 and RT in t1. The tube 0 is now filled with the solvent and the tubes are mixed.
After the mixing in the tube 0, the remaining fraction FR is now separated in the organic and the aqueous phase. In the aqueous phase (considering that the organic phase is transferred each time), there is now the remaining fraction of the first remaining fraction, i.e. FR.FR. In the organic phase, there is the transferred fraction of the remaining fraction, i.e. FR.FT.
In the tube 1, the transferred fraction from the tube 0 is separated between the two phases. In the aqueous phase there is now FR.FT and FT.FT in the organic phase. Once the mixing is done, the organic phases will be transferred to the next tube. The aqueous phases stay in their tube. FT2 is transferred from t1 in t2 and FR.FT is transferred from t0 to t1. After the transfer, we find respectively in the tubes 0, 1 and 2, FR2, 2FR.FT and FT2.
After three transfers, we would have FR3, 3FR2.FT, 3FR.FT2 and 3FR3. You may have noticed that those values are the members of an exponent of a sum:
![]()
n being the number of transfer. In a given tube r, the fraction is
![]()
We can thus predict the composition of each tube knowing the result of one single extraction. For a large number of extraction, we find the equation of a Gaussian curve.
The width of the peak is characterised by a standard deviation
![]()
The peak moves linearly with n, spread as √n and the intensity of the peak decreases as 1/√n.
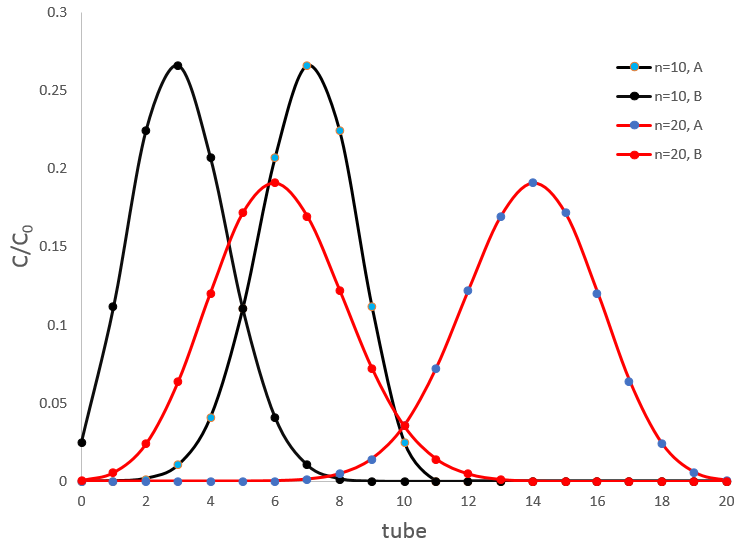
If two substrates are in the sample, the apparatus of Craig will separate them. Again, we have to choose carefully the conditions of the extraction to obtain a good separation. The figure above shows the concentrations of two species with DA=1/DB=9/4 after 10 and 20 transfers. There is an overlap between the curves where the species are not well separated but it decreases significantly with the number of extractions. At the maximum of the peak for A, after 10 transfers we find ~27% of the initial concentration of A but also 1% of the initial concentration of B. After 20 transfers, we have 19% of [A]0 and only 0.03% of [B]0.
The advantages of the apparatus of Craig is that everything is mechanical, so the precision is good and that we can either get the result of the extraction at the end of the apparatus or at one given tube in the apparatus.