Aromatic cycles, such as the benzene, are very stable because of their energy of resonance. As a result, it is very difficult to “open” the cycle by an usual reaction of addition.
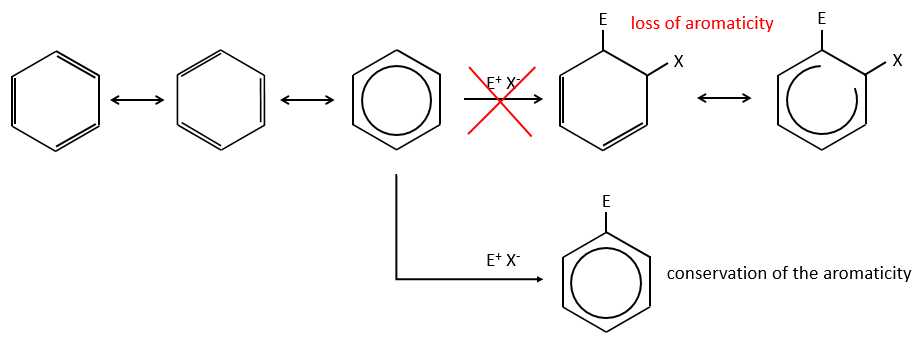
Instead of addition reactions, we observe reactions of substitution. The mechanism involves two steps.
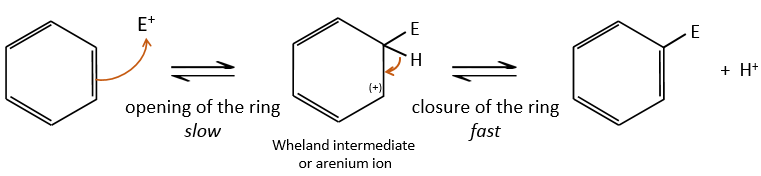
- the first step is the electrophile attack of one pi liaison on an electrophile. As this step implies the loss of aromaticity of the substrate, this step is slow and is the determining step of the reaction. The product of this step is called an intermediate of Wheland or an arenium ion.
- the second step involves the removal of a proton from the cycle to regenerate the aromaticity. This step is fast.
Most of the electrophiles have to be activated to allow the first step of the reaction. It is done by a Lewis acid.
Halogenation
FeBr3 and AlCl3 can be used as Lewis acids to bind a halogen (Br and Cl respectively) on a benzene ring.
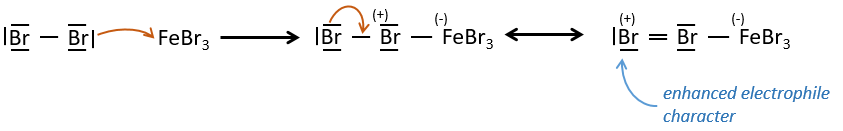
They enhance the electrophile character of the halogen that can now be attacked by the aromatic substrate.
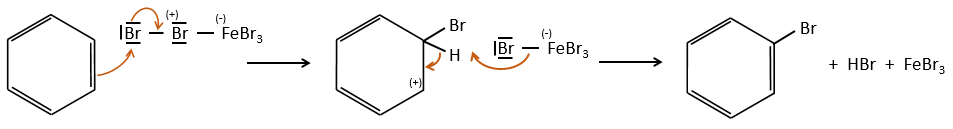
FeBr4– acts like a base to take a proton and close the ring.
In the following table, we can find the energies of liaison involved in the process for the halogens. On the left, we have the energies for the reactants and in the middle column we have the energies for the products. The enthalpy of reaction is shown on the right column.
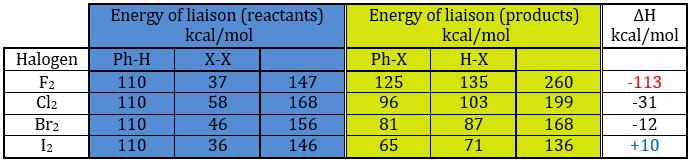
If we take a look at the variation of energy involved by a halogenation, we see that the enthalpy of reaction is positive for iodine. The reaction is thus not done. The reaction is highly exothermic for F2. In fact, this reaction is explosive. For Cl and Br, we need to use catalysts (the Lewis acids).
Sulfonation and nitration
SO3 and NO2+ are electrophile enough to bind with an aromatic ring.
Nitration
The nitrate can be reduced selectively to obtain an amine.
Sulfonation
The sulphur atom is electrophile because of the electrocaptor character of the oxygen’s. However, the reaction is reversible in presence of water to form sulphuric acid. This process is exothermic and we should keep an eye on it.
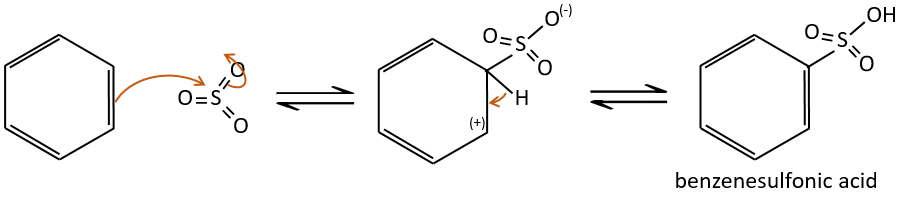
We produce detergents from the benzenesulfonic acid.
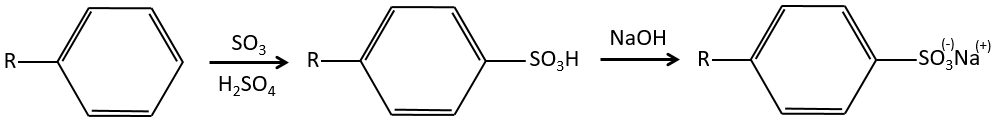
And from that point we can produce sulfonamides that are usually good antibiotics.
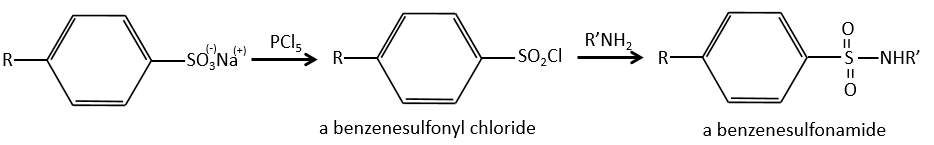
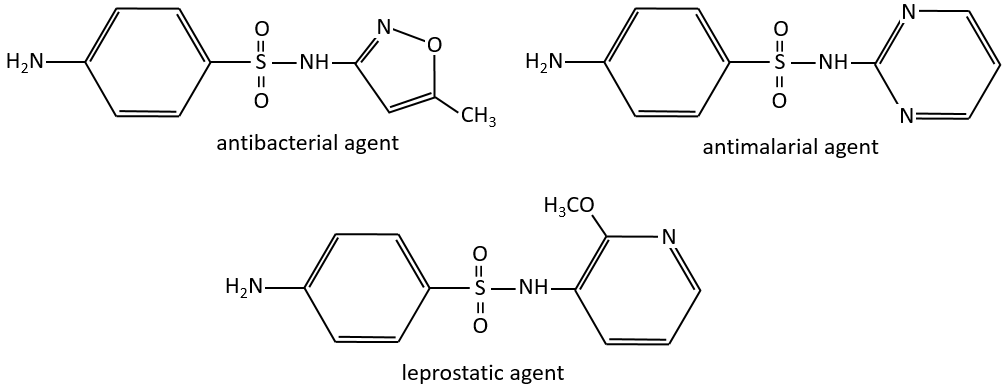
The first of this kind was the Prontosil (4-[(2,4-Diaminophenyl)azo]benzenesulfonamide) in 1932, developed by Domagk at Bayer, who won a Nobel prize in medicine for it in 1939. The research program was designed to find dyes that might act as antibacterial drugs in the body. The discovery and development of this first sulfonamide drug opened a new era in medicine. Other examples of antibacterial agents follow:
Alkylation of Friedel-Crafts
This reaction, as its name mentions, allows to bind a chain on an aromatic ring. The chain that is to be added has to have an electrophile carbon. Again, we need a Lewis acid to do this reaction. The first step is the activation of the halogenoalkane by the Lewis acid.
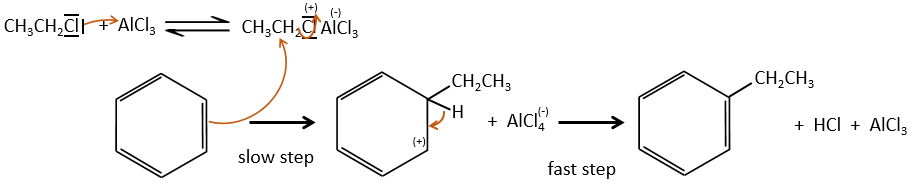
Next, the cycle attacks the alkane, ejecting the halogenated Lewis acid. The product of this step is called an intermediate of Wheland or an arenium ion. This step is the determining step of the reaction, involving the opening of the ring due to the electrophile attack. A proton is taken by the halogenated Lewis acid to give the aromaticity back with the recovering of the Lewis acid and the liberation of an acid. This step is fast with regard to the previous step. As a global result, we added a new carbon chain on the phenyl.
The halogen on the added chain is not mandatory. An alcohol can do the job as well, or other precursors of carbocations.
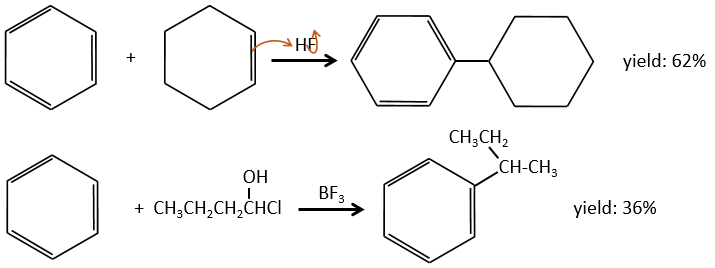
The alkylation can be intramolecular if it results in a new cycle.
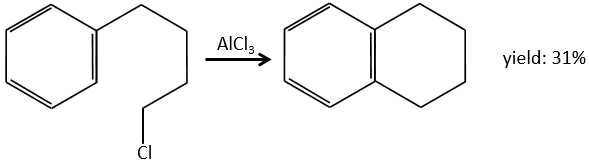
Limitations of the method
The alkyl group that has been added to the phenyl is an inductive donor group. It means that it gives some of its charge to the phenyl, increasing its ability to attack electrophilic carbons. As a result, the alkylation process does not stop after the addition of one chain if there is still places on the phenyl to greet new ones (we will see a bit later on which spot which group can be added).
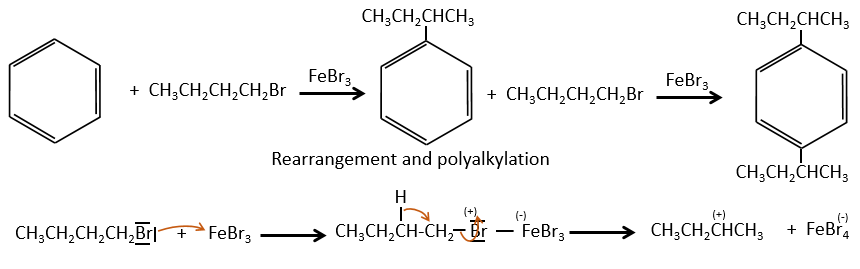
A second limitation is the rearrangement of the carbocation (as usual).
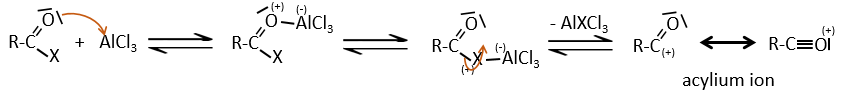
Acylation of Friedel-Crafts
The difference between the alkylation and the acylation is that for the second one the electrophile is an acylium cation, leading to the addition of -C=O-R on the phenyl.
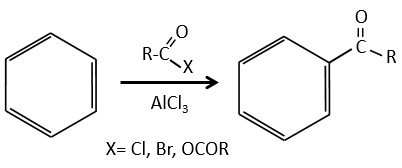
The acylium ion is obtained with the help on a Lewis acid
The rest of the process is identical.
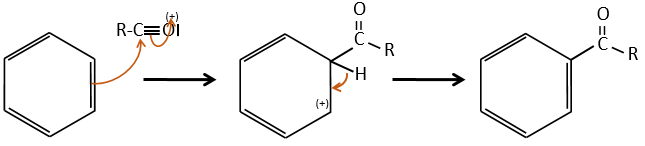
However, there is no polyacylation because the carbonyl is a mesomeric and inductive captor, taking electrons from the cycle. It is possible to reduce the ketone with the reaction of Clemmensen or of Wolff-Kishner.
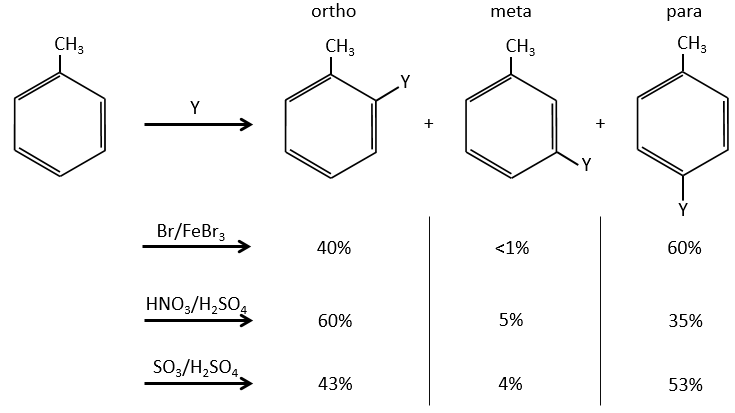
Regioselectivity
A benzene that already wears a group can be attacked on 3 non-equivalent positions: the ortho, meta and para positions. Some groups orient the reaction towards the ortho/para positions and other groups towards the meta position.
Mesomer effect
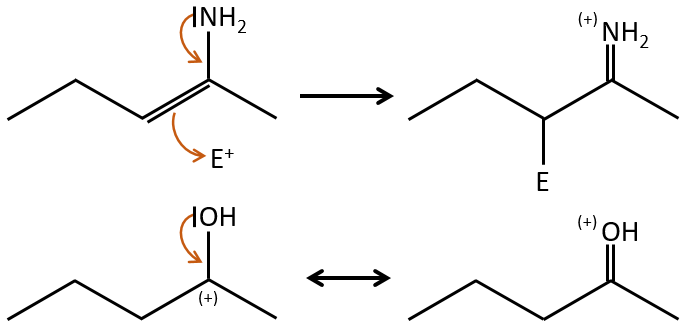
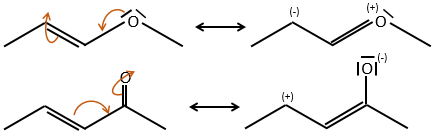
The mesomeric effect is the possibility for a heteroatom to share one lone pair with nearby atoms. For instance, O and N are mesomeric donors. They can stabilise carbocations by the sharing of one of their lone pairs that counterbalances the positive charge. They also enhance the nucleophile character of double liaisons. We note the mesomeric donors +M.
Groups can also be mesomeric acceptors and are noted –M. For instance the NO2 group is a mesomeric acceptor because it has a resonance form that can keep the additional charge.
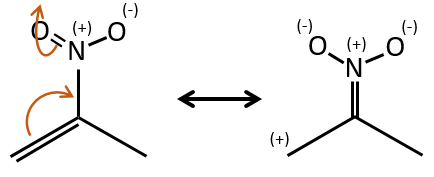
Note that the oxygen can be mesomeric donor or acceptor depending on the group it is in and of the structure of the molecule.
Inductive effect
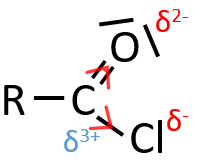
When two atoms are of different electronegativity, there is a displacement of electrons from the least electronegative atom to the most electronegative one. Groups or atoms can thus take or give some electrons from or to the nearby atoms. Atoms or groups that take electrons from the rest of the molecule are inductive acceptors and are noted –I. They destabilise carbocations and positive charge and can enhance the electrophile character of the atom they are bond with. For instance, the carbon of a carbonyl give a part of its charge to the oxygen and wears a partial positive charge δ+. A nucleophile is thus prone to attack this carbon.
An atom that is less electronegative than the carbon will give a part of its charge to the chain. They are called inductive donors and noted +I. The simplest inductive donor is H that is less electronegative than the carbon. However, H is taken by convention as the neutral intensity of the inductive effect. –CH3 is an inductive donor because the inductive effect of the 3 hydrogen’s is transmitted through the carbonic chain (over 2-3 carbons). As a result, the alkyl groups are inductive donors. The effect is stronger for -C(CH3)3 than for –CH3 because the amount of hydrogen atoms that can share their electrons.
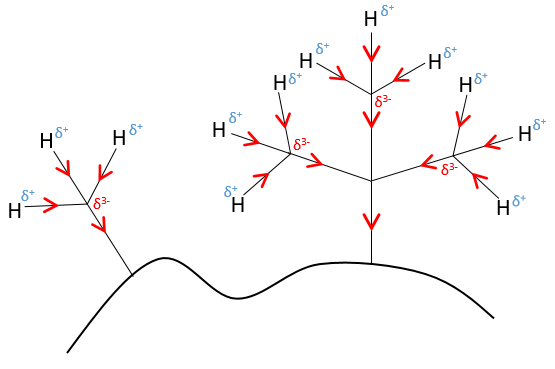
The hybridation of the carbon is something to be taken into account to determine the inductive effect. The electrons in the s orbitals are more bound to the atom than the ones from the p orbitals. As a result, the electronegativity of sp2 carbons is slightly higher than the electronegativity of sp3 carbons.
Regioselectivity in function of the substituents of aromatic rings
The mesomeric and inductive effects are cumulative and a nitrogen in a NH2 group is simultaneously a mesomeric donor and inductive acceptor (+I-M). The mesomeric effect always prevails in intensity.
- inductive donors
The groups that are donors of electron by hyperconjugaison (or inductive donors) are activating, i.e. favour the addition of new groups on the carbon. The substitution is oriented towards the ortho and para positions because the carbocation which has been made during the first step of the substitution can be stabilised by the inductive donor.
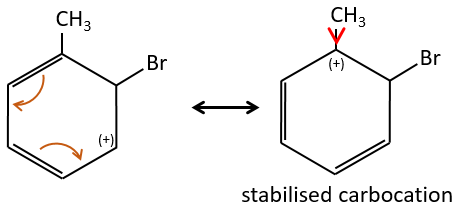
It is not possible to place the positive charge on the spot of the group if the substitution was to be made on the meta position. Usually, the para position is favoured to the ortho position because of the steric hindrance. However some groups such as NO2 seem to show more interest on the ortho position.
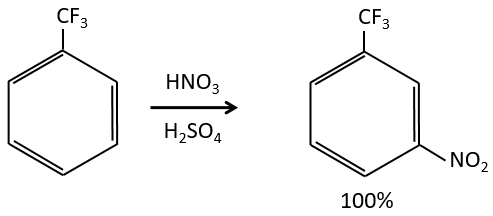
- inductive captors
Inductive electrocaptors are deactivating and orientate the reaction in meta. The reason is that in the other two positions, the carbocation can be at the feet of the CF3 group that wants to take more electrons, destabilizing furthermore the carbocation.
- mesomeric donors
Mesomeric electrodonors are activating and orientate the reaction in ortho/para because there is an additional resonance form.
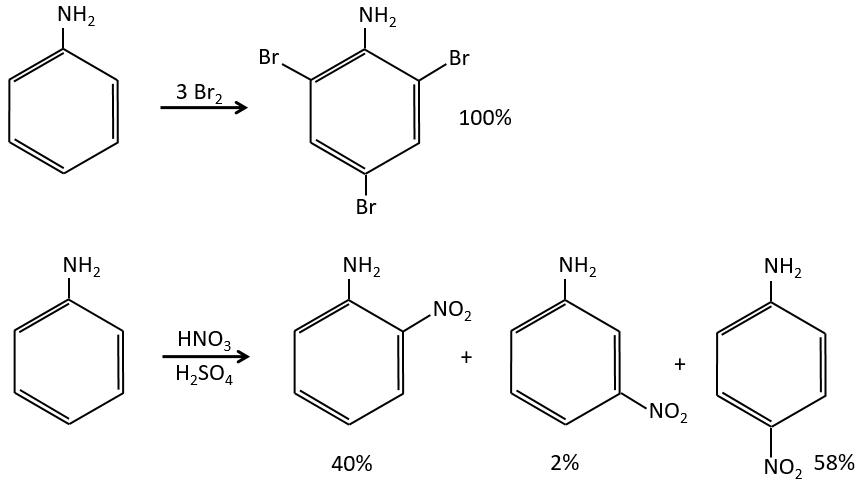
Note that NH2, as well as the oxygen from the ether, is a mesomeric donor and an inductive captor. The mesomeric effect is always more important than the inductive one.
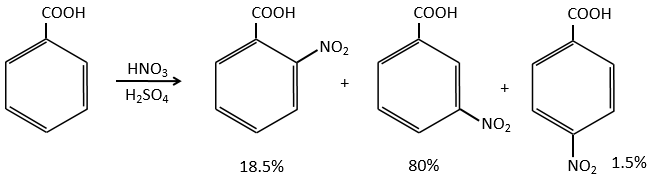
- mesomeric captors
Groups that are electrocaptor by resonance are deactivating and orient the reaction in meta.
- halogens
Halogens are deactivating but orient the reaction in ortho and para.
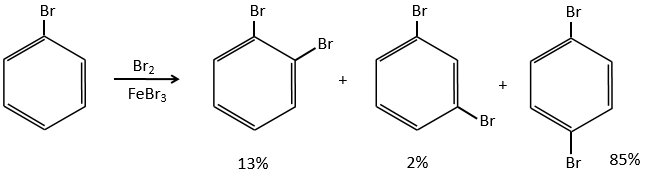
The steric hindrance here is very important and explain the difference of population between the ortho and para reactions.
If there were two groups on the phenyl, the effects are additive and the substitution is made on the most activated/ less deactivated positions.
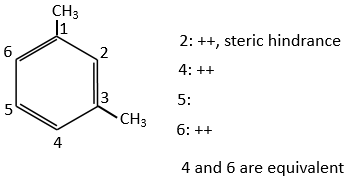
Both methyl groups of the xylene (dimehtylbenzene) shown above are activating and orientate in ortho/para. As both groups are activating, this reactant is more reactive than the toluene. The positions 2, 4 et 6 are thus favoured. The positions 4 and 6 are slightly more probable because there is less steric hindrance. In the case of the following xylene, the slots are almost equivalent.
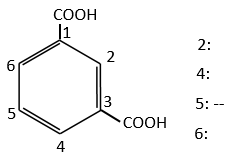
In the case of deactivating groups, the same reflexion is made. The COOH’s orientate in meta and the position 5 is the least deactivated position.
Nucleophilic substitutions
The nucleophilic substitution on aromatic rings is slower than the electrophilic substitution and that substitutions on sp3 carbons.
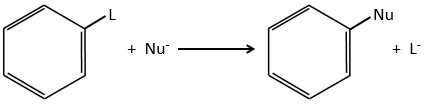
The reason is that the cycle is already full of electrons. Moreover, a sp2 carbon is more electronegative than a sp3 carbon. It is thus difficult to add a nucleophile (that loves positive charges) on a phenyl and the presence of a captor group on the phenyl is required to stabilise the carbanion. The leaving group has to be a good one.
Several mechanism are possible to add a nucleophile on an aromatic substrate. One of them is the Addition-Elimination.
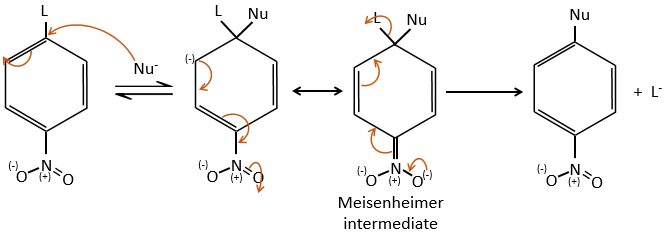
The activating group (NO2 here), is necessary to stabilise the negative charge of the carbanion formed during the first step of the reaction which is the slowest one. The intermediate complex is called intermediate complex of Meisenheimer.
The halogen (as leaving group) reactivity sequence is opposite to the one for SN2. On an aliphatic chain, the cleavage of the C-X liaison is made during the determining step of the SN2. On an aromatic, the cleavage doesn’t take place during the determining step. Moreover, the halogens take electrons from the cycle and the small halogens are thus more reactive than the big ones. As a result, we have the following reactivity sequences:
Aliphatic: F<<Cl<Br<I
Aromatic: F>>Cl>Br>I
A heterogeneous atom in the cycle (O or N) can play the role of the captor group.
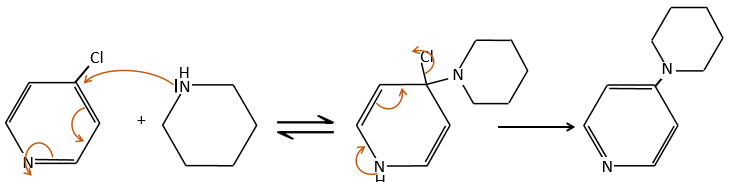
An application of this mechanism is the determination of the terminal amino acid of peptides. The peptide reacts with an aromatic compound through its terminal amine group.
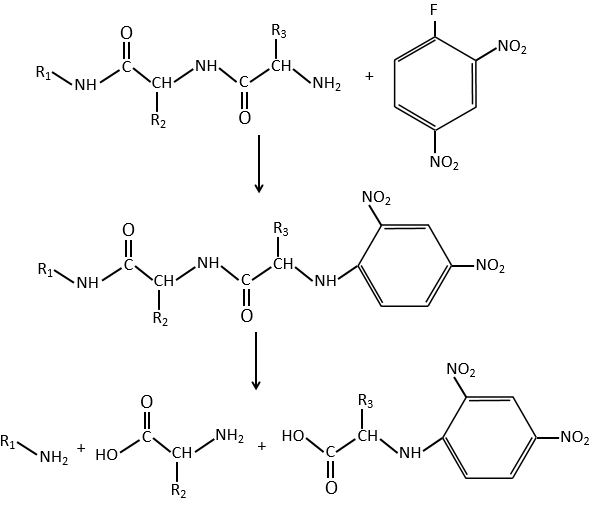
Once the two substrates are bound together, we hydrolyse the peptide bonds (i.e. the amides). All the amino acids are now separated but only one of them is bound to the aromatic group.
SN1 mechanism
This mechanism is mostly used to produce salts of arenediazonium. This species is obtained from aniline C6H7N with NaNO2 in an acidic environment.
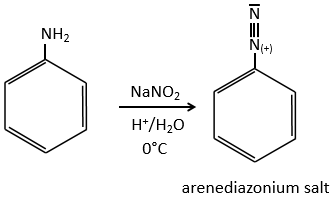
The mechanism is the following:
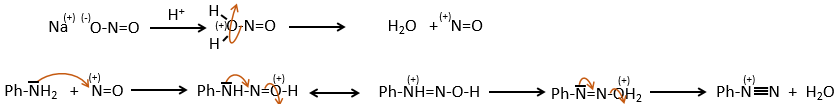
Arenediazonium salts are stables at low temperatures and lose N2 at higher temperatures, freeing a phenyl cation that can react with a nucleophile.
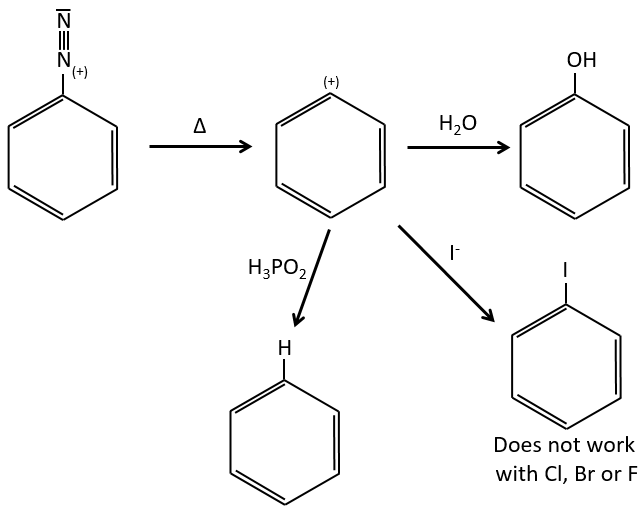
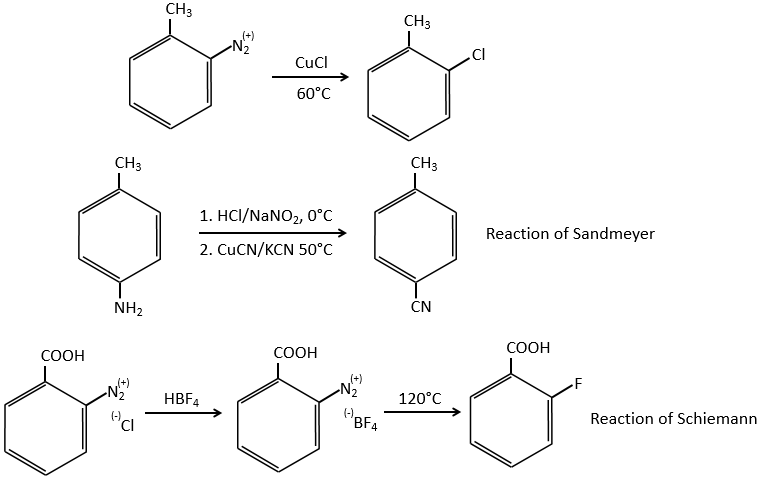
Other halogens than iodine don’t give good results because of secondary reactions. To add Cl, Br or F on an aromatic cycle, we use the Sandmeyer reaction, using copper salts like CuCl, CuBr or CuCN. The mechanism is a bit more complex and involves radicals.
Mechanism involving a benzyne
Normally, halogenoarenes cannot make usual SN2 or SN1 reactions. However, in very harsh conditions of temperature and of pH, it is possible to force such reactions.
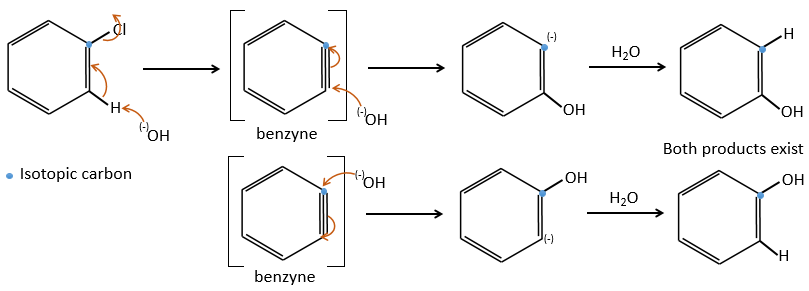
The mechanism involves a triple liaison in the cycle, the species being called a benzyne, The existence of this intermediate has been shown by an isotopic labelling. The C bound to the halogen is an isotope and we observe a racemic melange as product: