The interest of electrophile additions on C=C liaisons is to transform two sp2 carbons into two sp3 carbons and to add a chain or a group on the existing molecule.
As two carbons are involved in the reaction, the addition of the new group can give several products, with more or less (stereo)selectivity. Moreover, the transformation into sp3 carbons also introduces a regioselectivity that often has to be considered.
Consequently, we will discuss in this section the methods and the rules that have to be followed to obtain the desired product during the addition of an electrophile group on double or triple liaisons.
Catalytic hydrogenation
It is, in the principle, the easiest addition on a C=C liaison because there is no new group on the product. The sp2 carbons are reduced into sp3 carbons. Yet, this reaction is not spontaneous despite the fact that it is an exothermic reaction. The activation energy to break the H-H liaison is huge and a catalyst is required to perform the reaction.
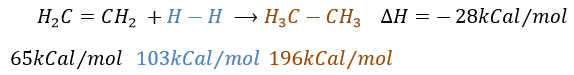
As catalyst, we can use platinum (Pt), Raney’s Nickel (Ni) and Palladium on C (Pd/C).
The have the ability to dissociate the hydrogen atoms and to fix them on their surface, making them available for the addition on the alkene. As detailed in the kinetics section, the catalyst decreases the energy of activation of the reaction and the process can be depicted in 4 steps:
– approach and binding of the reactants
– displacement on the surface
– reaction(s)
– departure of the products
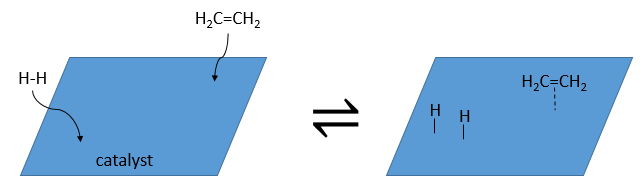
At the first step, the reactants take place on the surface of the heterogeneous catalyst. The amount of reactant that can take place on the catalyst depends on its surface. It is thus advised to use small particles of catalyst to increase the ratio surface/volume (or surface/mass). The alkene places itself parallel to the surface, in the direction of the π liaison.
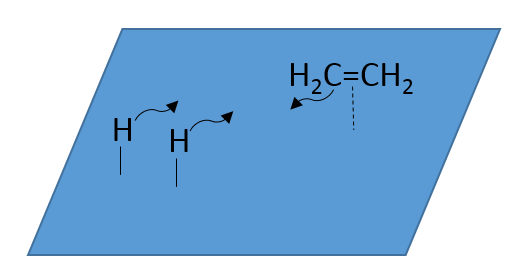
There is no reason that the reactants immediately bind on nearby spots. The second step is thus that the reactants are moving on the surface. They will eventually be in contact with each other so that the reaction can take place.
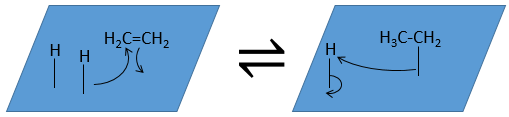
The third step (and eventually the next ones) is the reaction between the reactants. One activated hydrogen attacks one sp2 carbon while the π liaison is now connecting the second carbon with the platinum. The next step of the reaction is that the liaison between the carbon and the platinum attacks the remaining hydrogen.
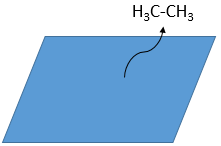
As last step, the alkane leaves the surface of the catalyst, letting space for reactants to bind.
The hydrogenation is stereoselective, i.e. the place of the groups on the carbons are not equivalent: at the level of the C=C liaison, the molecule is planar. If the hydrogen atoms are fixed on the same side (but not on the same carbon) of the molecule. It is a syn addition. As the added groups are hydrogen’s, the product is thus cis (see nucleophilic substitutions about cis and trans products).
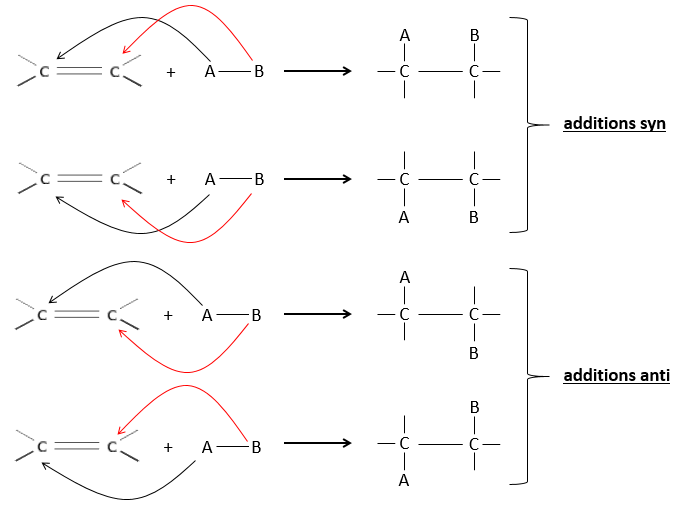
If the groups were added on the opposite sides of the molecules (still not on the same carbon), the addition would be anti and the product would be trans. This product is not observed because the reactants are bound to the catalyst and cannot move freely during the reaction. Both hydrogen’s are thus on the same side of the molecule before the reaction and after the reaction.
This is of importance only if, after the reaction, the newly sp3 carbon cannot rotate. It will be the case when the liaison is involved in a cycle or if there are voluminous groups. Moreover, the fact that we obtain only the cis product does not mean that only one product is possible. The reason is that there is one plane at each side of the π liaison (for a total of 2). If the two planes are equivalent, we will obtain a 50-50 racemic mix of two cis products.
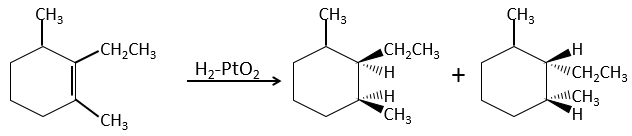
If the planes are not equivalent, one product is favoured: the hydrogen’s are fixed on the least occupied side because the alkene places itself on the catalyst so that the voluminous groups are far from the catalyst.
Soluble catalysts can also be used for this reaction. It is the case for the Wilkinson’s catalyst chloro(tris(triphenylphosphine)rhodium(I).
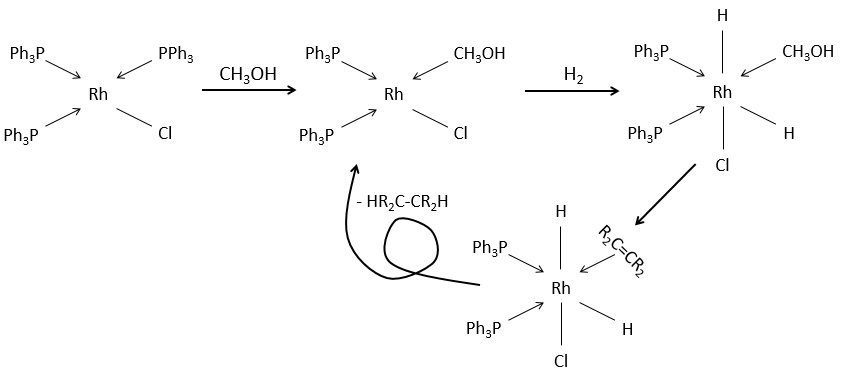
It also lead to the cis products.
In terms of reactivity, the substituted carbons react slower than carbons with less groups because of the steric hindrance: the hydrogen’s are not very mobile on the surface of the catalyst and the π liaison has to be nearby to obtain the hydrogenation.
The alkynes also react faster than alkenes, allowing us to hydrogenate all the alkynes into alkenes before the formation of alkanes. We can thus limit the hydrogenation to the alkenes if we use a bad catalyst and put one equivalent of H2 by alkyne. We will obtain the alkane with 2 equivalents of H2 by alkyne.
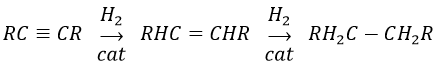
Again, it will lead to the cis alkene. To obtain the trans alkene, we use sodium in ammoniac. The sodium generates a radical on one carbon and a negative charge on the other one.
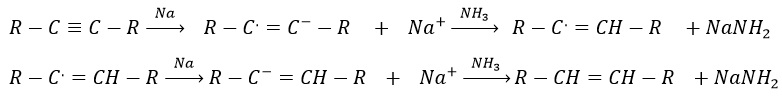
The trans product is favoured for steric reasons.
Aromatic products can difficultly be hydrogenated with H2/cat (at 150°C) but it can also be reduced differently by the reduction of Birch, involving Na, NH3 and an alcohol.
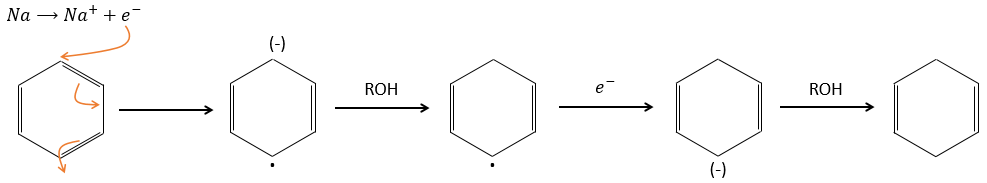
Other electrophile additions
Addition of HX
The other reactants HX containing a hydrogen atom can more easily be broken (or are already ionic) and do not require a catalyst. In the case of a halogen acid, because of their nucleophilic character, the π liaisons which are weaker than σ liaisons tend to attack electrophile molecules. In this case, the electrophile target is the proton. It is called an electrophile attack. The nucleophile X– attacks the positive carbon (or carbocation) afterwards.
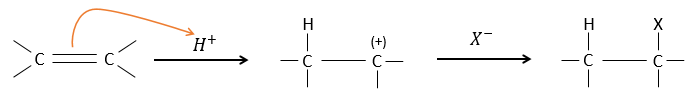
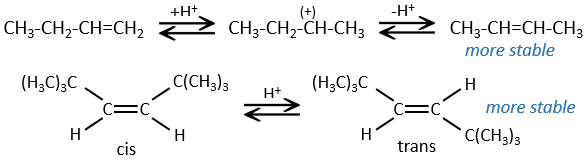
This reaction does not involve stereoselectivity because there are two distinct steps but involves regioselectivity: it is the rule of Markovnikov that says that the halogen is fixed on the most substituted carbon, or inversely that the proton is fixed on the least substituted carbon. Personally, I prefer to remember that the most stable carbocation is formed. Remember that carbocations can rearrange the molecule to form the most stable carbocation.
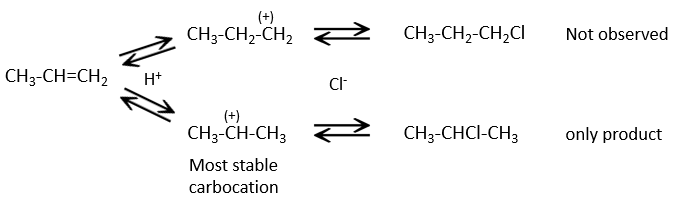
Addition of water
Water is added to alkenes in presence of sulphuric acid.
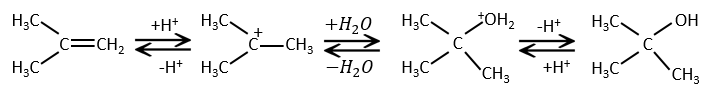
The sulphuric acid is chosen because the sulphate is less nucleophilic than the water because the charge is stabilised by resonance. HCl has to be avoided because Cl– is more nucleophilic than H2O. Moreover, all the steps are reversible. It means that the newly generated alcohol can be dehydrated in the case of an excess of acid.
The presence of a carbocation explains some rearrangements of alkenes that can be observed in acidic solvents.
Addition of X2
Halogen’s are added to C=C to obtain trans products.
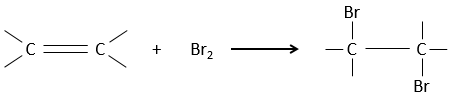
It is strange that something as charged in electrons as halogens are used as electrophiles for this reaction. The reason is that the liaison between the halogens is polarisable. A halogen will thus feel the electrons of the π liaison and transfer part of its electrons to the other one. The partially positive halogen can thus be attacked by the π liaison and forms an intermediate halonium that will be attacked by the negative halogen on the other side of the C-C liaison.
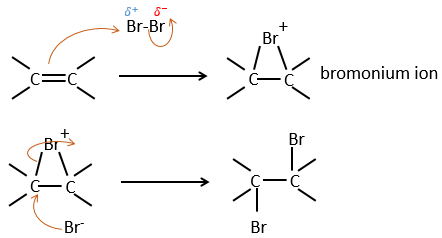
This reaction is stereoselective (always trans) and stereospecific (the result depends on the arrangement of the reactant). In presence of other nucleophiles, such as water, the reaction begins normally but there is a competition between X– and the nucleophile.
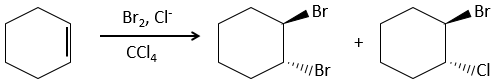
This reaction is slower on alkynes because the intermediate halonium is less stable. The halogenation is thus hard to control: the alkenes will react faster than the alkynes.
The oxymercuration reaction
This reaction is a useful way to form alcohols or ethers from alkenes. It involves mercurial acetate and water/alcohol in THF as solvent. The nucleophilic opening of the water/alcohol is done in anti and on the most substituted carbon (so there is more space for the mercurial acetate).
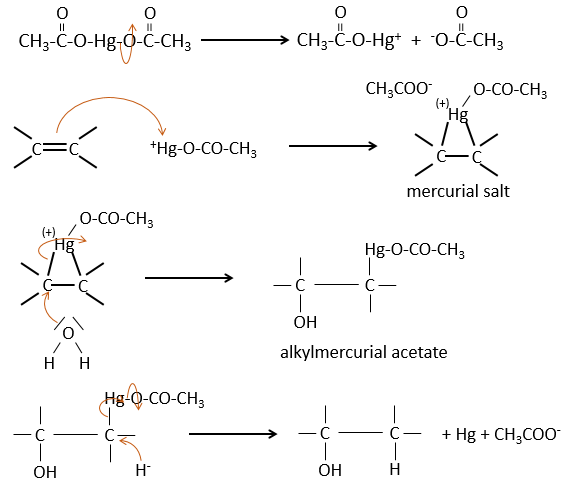
The oxymercuration follows the Markovnikov rule but there is no carbocation involved in the process. As all the additions, it is regioselective.
Water leads to the formation of an alcohol and we can use an alcohol instead of the water to obtain an ether.
Hydroboration reaction
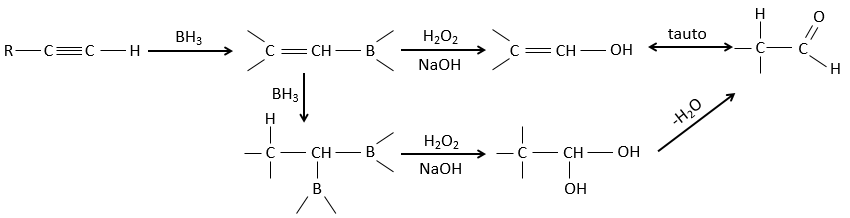
BH3 (borane) is a Lewis acid that is stable under the form of a dimer and can replace its 3 hydrogen by 3 carbon chains in THF (tetrahydrofuran (CH2)4O). Once the chains are bound, it is possible to remove the boron by an oxidation with H2O, NaOH and H2O2 to obtain as many alcohols.
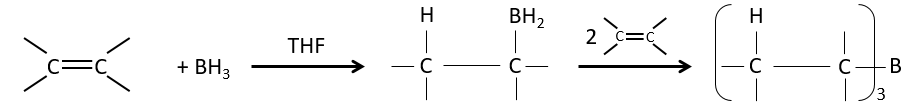
This reaction does not follow the Markovnikov rule (anti-Markovnikov) and is stereoselective. The mechanism of reaction involves the empty p orbital of the boron that interacts with the π liaison to form a complex. This complex allows a transition state with 4 centres wherein the liaisons move.
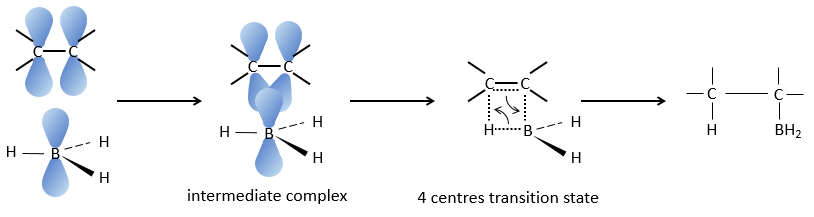
The Markovnikov rule applies when the hydrogen is positively charged. In the present case, we add H– and BH2+.
The removal of the boron is made with retention of configuration and is stereospecific. The boron is less electronegative than the carbon and is thus giving its electrons. NaOH takes a proton from the H202 to form HOO– that attacks the boron, transferring its negative charge. There is a rearrangement occurring that puts an oxygen between the chain and the boron.
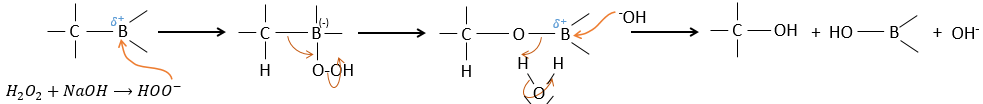
The water and OH– take action next to remove the boron.
This addition can also be done on alkynes to form aldehydes through tautomerization.
Reaction of alkenes with peroxycarboxylic acids
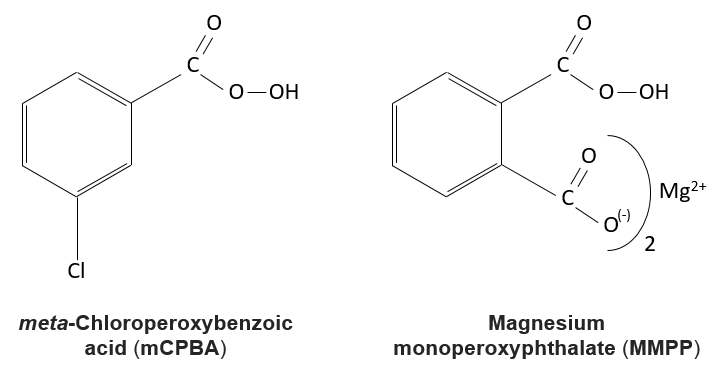
The peroxycarboxylic acids are generally unstable. The MCPBA and the MMPP are however stable peroxyacids vastly used in laboratory.
One of the oxygen of the peroxycarboxylic acids is electrophile. Those acids can thus react with alkenes to form epoxides and the carboxylic acid.
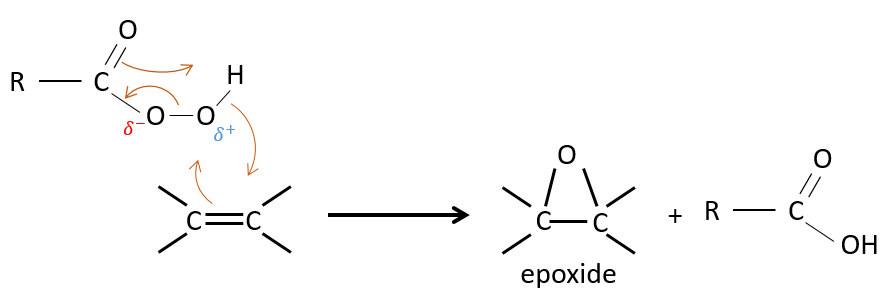
The addition is thus a concerted reaction, is stereospecific and is syn. The reaction is made on the less hindered plane of the alkene. The reactivity increases with the amount of substituents that share their electrons with the π liaison (alkyl chains for example).
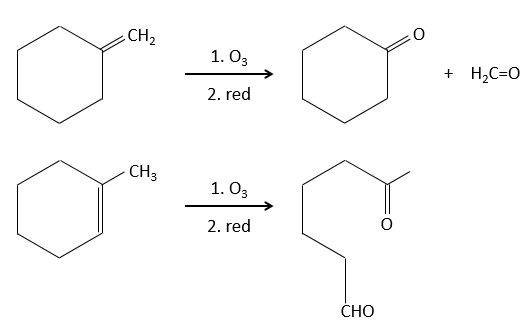
Ozonolyse
The ozone can break double liaisons. As a first step, it opens the double liaison to give the intermediate called molozonide.

It breaks to form ozonide that can next be oxidized or reduced.
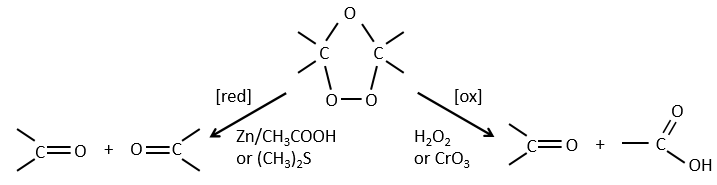
The following table gives the products of the ozonolyse as a function of the substituents of the double liaison.

The ozonolyse is thus a good way to obtain ketones and carboxylic acids from alkenes. The method is also useful to determine to position of a double liaison in a molecule when we cannot easily determine it with usual techniques.