The elements of the copper-arsenic group are Hg2+, Pb2+, Bi3+, Cu2+, Cd2+, As3+, Sb3+ and Sn4+. They form sulphides that are insoluble in dilute HCl. It is not the case for sulphides of the following groups so we can isolate the ions from the copper-arsenic group from the ions of the other groups. Lead and mercury were already in the Silver group but they appear in the copper-arsenic group as well. Lead chloride is somewhat soluble in dilute HCl so the ions may not be completely precipitated in the silver group. Mercury has two oxidation states. Hg2Cl2 is insoluble in dilute HCl but HgCl2 is very soluble. As a result, mercury(II) is not precipitated in the silver group.
The sulphides are formed with the weak acid H2S. Example with Hg2+:

The metal sulphide begins to precipitate when the product of the concentration of the cation and the sulphide ion equals the solubility product for the sulphide and the precipitation continues until it is not the case anymore. It is an equilibrium. The pH of the solution plays a role: the two first steps of the reaction are the deprotonation of H2S. If we increase the pH of the solution by the addition of HCl, the equilibrium
![]()
is shifted to the left and the S2- concentration decreases. On the contrary, if we dilute the solution but keep it saturated in H2S, the concentration of the S2- ion increases. We want thus a small H+ concentration to have a large concentration of S2-. The H+ concentration can be decreased by dilution or by addition of the salt of a weak acid (NH4C2H3O2 for instance, NH4+ replaces H+ in the solution with a negligible effect on the pH of the solution).
Two factors modify the application of the rule that we want a high S2- concentration and thus an H+ concentration as low as possible. Arsenic, usually present as arsenate, is converted into As2S5. To obtain it, the reaction requires a high concentration of H+. At the other extreme, if the acidity is too low, the sulphide ion concentration will be so high that the solubility products for the sulphides of iron, zinc, manganese, cobalt and nickel (not in the copper-arsenic group) will be exceeded and will also be precipitated.
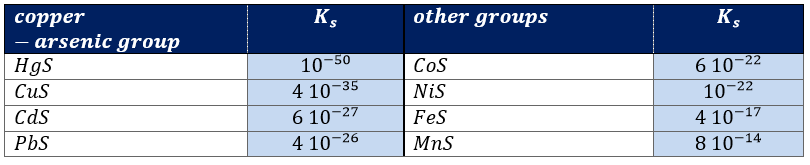 Let’s now take a look at the ions of the copper-arsenic group.Due to the successive dilutions we make in the experiments, the Co2+ concentration should not be larger than 0.02M, so the S2- concentration should not exceed 3 10-20M. The overall ionisation constant for H2S is 1.3 10-20. Since the solubility of H2S in water at room temperature is about 0.1M, the value of the ion product [H+]2[S2-]=1.3 10-21, meaning that if we want to keep the S2- concentration lower than 3 10-20M, [H+] must be no lower than about 0.2M. So, even if the analyses are not quantitative, we have to be careful to the quantities we use in this procedure.
Let’s now take a look at the ions of the copper-arsenic group.Due to the successive dilutions we make in the experiments, the Co2+ concentration should not be larger than 0.02M, so the S2- concentration should not exceed 3 10-20M. The overall ionisation constant for H2S is 1.3 10-20. Since the solubility of H2S in water at room temperature is about 0.1M, the value of the ion product [H+]2[S2-]=1.3 10-21, meaning that if we want to keep the S2- concentration lower than 3 10-20M, [H+] must be no lower than about 0.2M. So, even if the analyses are not quantitative, we have to be careful to the quantities we use in this procedure.
Description of the elements of the silver group
Mercury(II)
It is the highest common oxidation state of mercury in aqueous solution. Aqueous solution containing mercury are poisonous. The soluble forms of Hg2+ are the halides, acetate and cyanide. The nitrate dissolves in dilute acids. Stable complexes are formed between Hg2+ and the halide ions or sulphide ions but do not bind well with NH3 and OH–. Few mercury(II) compounds are coloured.

Lead(II)
Lead(II) was already discussed in the previous section.
Bismuth(III)
It is the most common oxidation state of bismuth in aqueous solutions. The free ion is strongly hydrolysed, forming insoluble basic salts. The following equation represents the hydrolysis of BiCl3:
![]()
Solutions containing Bi3+ must be highly acidic to keep the cation in solution. In water, a milky white suspension may result from the formation of the insoluble white basic salt of bismuth and antimony. Bismuth can be oxidised to the 5+ state (ex: NaBiO3) by very strong oxidizing agents and is sufficiently powerful to oxidize Mn2+ to MnO4-.
The majority of bismuth compounds are colourless (exception: brown Bi2S3) Complexes can be formed in concentrated solutions of halides: Bi2S3 dissolves in 12M HCl.
Copper(II)
There are two oxidation states for the copper: 2+ is the most common and 1+ the second possibility. Cupric chloride, nitrate and sulphate are soluble in aqueous, and aqueous solutions of Cu(II) salts are blue, the colour of the always present complex ion Cu(H2O)42+. Most Cu(II) solids are also blue (hydrates) and contain this ion. Other usual ligands are NH3, Cl– and CN–.
All Cu(I) salts are insoluble in water and are colourless solids (exceptions: Cu2O (red) and Cu2S (black)). They are often precipitated from the reduction of Cu(II) salts. For example,
![]()
Cu(I) forms stable complexes with halide and cyanide ions and with ammonia, complexes oxidized in solution by atmospheric oxygen.
Cadmium(II)
The only common oxidation state of cadmium in aqueous systems is 2+. Halides, nitrate, sulphate and acetate of cadmium are soluble in water. The salts are colourless (exceptions: CdS (yellow), CdO (yellow brown). Cadmium forms complex ions with common ligands (halides, NH3 and CN–).
Arsenic
It exists in either the 3+ or 5+ oxidation states. It is usually not present as its ion but as an arsenate (AsO43-) or arsenide (H2AsO3–). If AsCl3 or AsCl5 are dissolved in water, they immediately hydrolyse to give the weak acids according to the equations
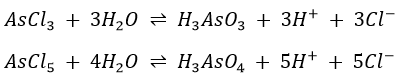
Sulphides are then precipitated.
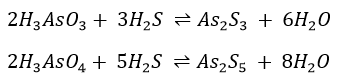
These reactions are slow but are speeded up when [H+] is increased. The amount of As2S3 formed by this series of reaction is small and can be oxidized to As(V) by (NH4)2S2 (procedure 6). We will thus not obtain any As(III) at the end of the process.
Antimony(III)
Antimony exists in the Sb3+ and Sb5+ states in its common compounds. The salts hydrolyse in water to form basic salts that are often insoluble. For instance,
![]()
However, antimony(III) tends to form soluble complex ions with halide or hydroxide ion. For example, SbCl3 is not soluble in water but will dissolve in hydrochloric acid solution, forming SbCl63-, or in bases forming Sb(OH)63-. The majority of antimony compounds are colourless (exceptions: SbS3 (red) and SbS5 (yellow)). In water, a milky white suspension may result from the formation of the insoluble white basic salt of bismuth and antimony.
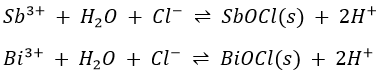
If treated by HCl, the mildness disappears because the equilibrium is shifted to the left. In concentrated HCl solution, antimony exists as the complex, stable tetrachloro-antimonate(III) ion, SbCl4–.
![]()
The hydrolysis stakes place when a strongly acid solution of antimony chloride is added to water.
![]()
Tin (II) and (IV)
Pure compounds containing Sn(II) and Sn(IV) are common. In aqueous solutions, both are stable but Sn(II) is slowly converted into Sn(IV) by air oxidation. Both tin oxidation states readily undergo hydrolysis to yield stannite’s (Sn(II)) or stannate’s (Sn(IV)). Thus common tin salts are insoluble in water because the hydrolysis products are insoluble basic salts such as SnOCl2.
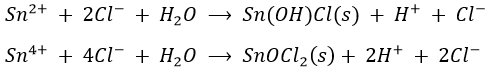
Tin compounds are usually colourless (exceptions: SnS (brown) and SnS2 (light yellow)). Both oxidation state forms complex ions, with the usual halide and hydroxide ions. As a result, although tin (IV) salts undergo hydrolysis to form insoluble salts, it forms the soluble, complex ions SnCl62- and Sn(OH)62- in hydrochloric acid and strong bases. The Sn(IV) hydroxide complexes are sufficiently stable to cause SnS2 to dissolve in 6M NaOH.
Procedure 5
Place the decantate from Procedure 1 in a casserole or in a lens and add two drops of 3% H2O2 and two drops of 2M HCl. Carefully boil down to a volume of 1 or 2 drops and then allow to cool. Add 6 drops of 6M HCl. Carefully evaporate the contents of the casserole down to a pasty mass (do not bake the residue). Cool and then we can either use H2S gas as the precipitating reagent (A) of a thiocetamide solution (B). To the residue of the casserole, add exactly 4 drops of 2M HCl, Swirl until all the residue is dissolved. If necessary, stir the mixture and warm slightly. Transfer the solution to a test tube.
(A) Heat carefully until it shows signs of effervescence and then treat with H2S under the hood for 20 to 30s.
Dilute with 10 drops of hot water and continue to treat with H2S for another 20-30s.
Add 1 drop of 1M NH4C2H3O2 and treat with H2S for another 20-30s.
Finally, add 25 drops of cold water and treat with H2S for another 20-30s.
Each time, make sure that H2S is bubbled all the way to the bottom of the solution. Centrifuge and test for complete precipitation by first noting the appearance of the solution and then passing H2S into the top of the supernatant liquid (do not disturb the precipitate). If precipitation is not complete, add 5 more drops of water and treat with H2S for an additional 20-30s. Centrifuge and repeat until the precipitation is complete. When it is the case, wash down the walls of the test tube with 2 or 3 drops of water, centrifuge and decant into a casserole. Boil it for one minute and save it for the analyses of the next groups (Procedure 15). Wash the precipitate twice with 15 drops of hot water and pass to Procedure 6. If the wash water peptizes the precipitate (makes a colloidal suspension from it, that does not settle when centrifuged), then add 10 drops of 1M NH4C2H3O2, mix well and heat nearly to boiling before centrifuging.
(B) Add 4 drops of 1M thioacetamide solution, mix thoroughly and heat in a boiling water bath for four minutes. Add then
– 8 drops of hot water,
– 8 drops of 1M thiocetamid and
– 1 drop of 1M NH4C2H3O2.
Mix well and heat in the boiling water bath for 4 minutes. Centrifuge and decant into a test tube. Save both the precipitate and the decantate. Test the decantate for complete precipitation by adding two more drops of 1M thioacetamide, mixing well and allowing to stand for 1 minute. If a precipitate forms (the previous precipitation was not complete), add two drops of hot water and two drops of 1M thioacetamide. Mix well and heat in the boiling water bath for 2 minutes. When a test shows that precipitation is complete, transfer the decantate to a casserole. Boil the decantate for 1 minute and save it for the test for next groups (Procedure 15).
Precipitates from the precipitation and from the tests for complete precipitation are combined by using a few drops of water to flush the latter into the test tube containing the first precipitate. Wash the precipitate 3 times: first with 10 drops of hot water and then twice with 20 drops of hot solution prepared using equal volumes of hot water and of 1M NH4C2H3O2. Analyse the content of the tube according to Procedure 6. As for (A), if the wash solution leads to a colloidal suspension, add 10 drops of 1M NH4C2H3O2 to the suspension, mix well and heat nearly to boiling, centrifuge, saving the precipitate for Procedure 6.
Notes
- In Procedure 5, the initial H+ concentration is 2M. In the course of the precipitation, about a tenfold dilution occurs, which should reduce the concentration to 0.2M. However, H+ ions are liberated during the precipitation. For instance,
![]()
As a result, the final H+ ion concentration is slightly higher than 0.2M. It ranges from 2M to 0.2M, which allows for complete precipitation of the copper-arsenic group without precipitation of the cations of the aluminium-nickel group.
The sulphides of mercury, lead and copper are black. SnS and Bi2S3 are dark brown but look black when wet. CdS, SnS2 and As2S5 are yellow. Sb2S3 is orange.
Lead may first form an orange precipitate of PbS.PbCl2 that changes to black PbS on continued H2S treatment. Mercury first precipitates as white HgS.HgCl2 and changes of colour through yellow, orange and brown to black HgS. The intermediate colours results from a mixture of the two precipitates in varying proportions. CuCl2 is green in concentrated solution and blue in dilute solution. The initial colour of the precipitates is thus not a good indication of the ion. The absence of any green of blue colour in an unknown solution shows the absence of copper.
2. If only members of the copper-arsenic group are present, we can use a different procedure which saves considerable time. Place 4 drops of the solution in a test tube. Add 6 drops of H2O2 and 10 drops of ammonium sulphide solution. Then add more ammonium sulphide until the solution turns yellow (about 30 drops). Stir the content of the tube.
Heat for 3-4 minutes in the boiling water bath while stirring. Avoid to heat to the point where excessive frothing of the contents occurs. Centrifuge and decant into a test tube. The decantate may contain AsS43-, SbS33- and SnS32- for Procedure 11. Repeat the treatment of the precipitate with a second 10-drop portion of ammonium sulphide solution, heating and stirring for 2 minutes. Centrifuge and add the decantate to the first. Save it for Procedure 11. Wash the precipitate twice with 20 drops of a hot solution prepared by mixing equal volumes of water a 1M NH4C2H3O2 and analyse the precipitate, which may consist of the sulphides of mercury(II), lead, bismuth, copper and cadmium (Procedure 6).
3. Evaporations in casseroles are made as follows: hold the casserole in your hand and pass it back and forth over the top of the flame of a Bunsen. At the end of every two or three back-and-forth passes, tilt the casserole slightly so that the solution will run to its lower edge. Be careful not to overheat to the point where the residue begins to bake. If brown areas develop on the bottom of the casserole, indicating baking, then swish the remaining solution around until the brown area is removed. When only 2 or 3 drops of liquid remain, remove from above the flame and let the heat of the casserole complete the evaporation. Baking may sublime off the chlorides of arsenic, mercury and tin.
4. Four drops of 6M HCl are added to the residue in the casserole before dryness in order that nitrate ions may be reduced in accordance with the equation
![]()
If present, nitrate ions will oxidize the H2S and precipitate S:
![]()
Other oxidizing agents may be present in a solution to be analysed and will react with H2S in acid solutions.

Fe3+ will not be affected by evaporation with 6M HCl but Cr2O72- is reduced:
![]()
5. To treat a solution with H2S, attach a clean glass bubbling tube to the rubber outlet tube at the source of H2 This bubbling tube is made by drawing down a glass tube of suitable diameter to a fairly fine constricted end. The overall length of the bubbling tube should be about 5in. Insert the end of the bubbling tube from which H2S is escaping into the surface of the solution in the test tube and then gradually bring it down to the bottom of the solution. The constricted tube will deliver a stream of very fine bubbles; large bubbles would tend to throw the solution out of the small test tube. If the tip of the bubbling tube is brought suddenly all the way to the bottom of the solution, the sudden rush of gas may throw the solution out of the test tube. A very rapid rate of H2S bubbling should be avoided.
An ordinary Kipp generator, placed in a hood, is a very satisfactory source of H2S. A trap, in the form of a bottle or flask should be placed between the generator valve and the point where the bubbling tube is attached.
One of the most satisfactory sources of H2S is the commercial mixture of sulphur, hydrocarbon mix, and asbestos, of which ”Aitch-Tu-Ess” is an example. This mixture can be purchased in bulk or in small capsules (in bulk is recommended). It is used for generation of H2S as follows. Set up a generator under the hood, as illustrated below.
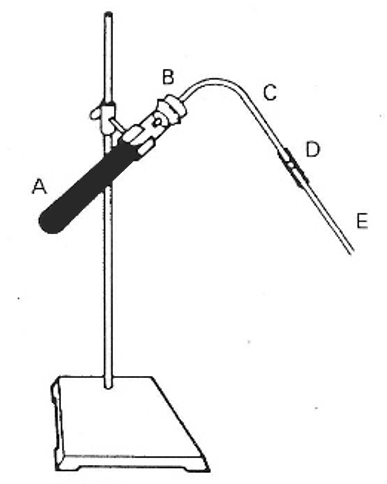
Fill the 6in tube A ½ to ¾ full of H2S-generating mixture. Insert the rubber stopper B, to which is attached the delivery tube C, the rubber connecting tube D, and the clean bubbling tube E, which is inserted into the solution to be treated. The heating should be gentle, just strong enough to give a fairly rapid evolution of H2S bubbles. The burner with which A is being heated can be held in one hand, while the test tube containing the solution is being held in the other hand. Do not heat so strongly that sulphur is distilled over. The evolution of H2S will cease when heating is stopped. The tube E should be removed from the solution being treated as soon as heating is stopped to avoid solution being drawn from the test tube up into E by the contracting gas.
The tube E should be removed and cleaned after each series of precipitations. Components A, B, C and D can be kept together when not in use. When the mixture no longer yields H2S on heating, tube A can be replaced by new mixture.
Hydrogen sulphide is a poison. Therefore all H2S treatment should be carried out under a good hood. Hydrogen sulphide is insidious since the sense of smell may become fatigued and fail to give warnings of high concentration. Low concentrations may produce irritation of the mucous membranes. Headaches, dizziness, nausea, and lassitude may appear after exposure.
6. Care should be taken so that the tube is not heated to the point at which excessive frothing results in loss of solution by spillage. If the tube is grasped with a test-tube holder, it can be withdrawn from the boiling water if and when spillage is imminent.
7. The organic compound thioacetamid, CH3CSNH2, hydrolyses in water (particularly at higher temperatures) to yield H2S:
![]()
It therefore serves as a convenient source of H2S. The hydrolysis is so slight at room temperature that a 1M solution of the compound, when preserved in a stoppered bottle, undergoes very little deterioration. At temperatures of about 80°C, the hydrolysis is sufficient so that a 1M solution yields a solution saturated with H2S.
Separation of the copper subgroup from the arsenic subgroup
We can separate the two groups in a solution of ammonium sulphide: sulphides of arsenic, antimony and tin(IV) are soluble and sulphides of mercury, lead, bismuth, copper and cadmium are insoluble.
As a general rule, the water-insoluble salt of a weak acid will dissolve in a strong acid. By this rule the metal sulphides should dissolve in HCl. The fact that they do not dissolve means that their normal solubility in water is so small that not even a strong acid can give a concentration of H+ sufficiently high to cause them to go into solution. For instance, the solubility of CuS is very low (4 10-35). It means that the concentration of sulphide ions in the equilibrium
![]()
is extremely low. The equilibrium reaction
![]()
which is set up when a strong acid is added to the sulphide, is not capable of giving a sulphide ion concentration lower than that in equilibrium with CuS and Cu2+. Consequently, CuS does not dissolve. On prolonged boiling with HCl, CuS will dissolve, very slowly. In this case the S2- ions are actually removed from solution as H2S. It should be noted at this point that the solubility principle does not tells at which speed an insoluble substance dissolves.
Ammonium sulphide ((NH4)2S) can dissolve As2S3, As2S5, Sb2S3 and SnS2, but not SnS. Therefore, if tin is in solution we need it as SnS2, thus present as Sn(IV) during treatment with H2S. To guarantee it, the solution is boiled with H2O2 before precipitation with H2S (see Procedure 5).
The separation of arsenic, antimony and tin from mercury(II), lead, bismuth, copper and cadmium depends on the fact that As2S3, As2S5, SnS2 and SbS3 are soluble in a solution of (NH4)2S, whereas HgS, PbS, BiS3 CuS and CdS are insoluble. To accelerate the process, the content of the tube in Procedure 6 are kept hot.
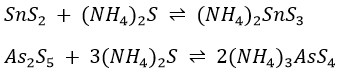
The soluble compounds are strong electrolytes and ionize to give NH4+ ions and SnS32-, AsS33-, AsS43- and SbS33- ions. The formation of the latter ions can be represented by the net equations
These ions may be classed as complex ions. Mercury(II), lead, bismuth, copper and cadmium do not form complex thiol ions. An examination of the formulas for the thiol salts suggests why these sulphides can be dissolved by (NH4)2S: (NH4)2SnS3 is the salt of thiostannic acid H2SnS3. It is the analogue of the ternary salt (NH4)2SnO3 in the oxygen system. Any ternary salt in the oxygen system may be considered to have been formed as a result of the reaction of a basic oxide with an acidic oxide. The oxides of metals are basic oxides, the oxides of nonmetals are acidic oxides.
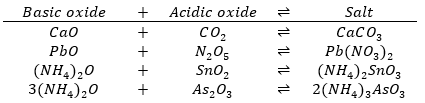
The sulphides of the elements bear the same relationship to HS that the oxides bear to H2O.
Therefore, CuS, PbS, HgS, CdS, SnS, Bi2S3 and (NH4)2S are basic sulphides in the sulphur system. SnS2, As2S3, As2S5 and Sb2S3 are acidic sulphides in the sulphur system. Basic sulphides react with acidic sulphides to form a salt in the sulphur system. Therefor we expect the acidic sulphides to react with (NH4)2S to form salts and HgS, PbS, Bi2S3, CuS, CdS and SnS should not react.
There remains the question of why SnS is basic while SnS2 is acidic. It is generally true that as the oxidation number of a polyvalent element increases, it becomes more non-metallic in character, and as an element becomes more non-metallic in character, its compounds become more acidic. Oxidation with H2O2 guarantees that the tin is present in its higher oxidation state.
Antimony(V) is very unstable and does not ordinarily exist in solution.
Procedure 6
To the test tube containing the precipitate from Procedure 5, add 10 drops of ammonium sulphide solution. Stir the contents of the tube well; then heat for 3-4 min in the boiling water bath, stirring the contents meanwhile. Avoid any excessive frothing of the content. Centrifuge, decant in a test tube and save the decantate which contains any present species of AsS43-, SbS33- and SnS32- for Procedure 11. Repeat the treatment of the precipitate with a second 10-drop portion of (NH4)2S, heating for 2 minutes. Combine the decantates together. Wash the precipitate twice with 20 drops of a hot solution prepared by mixing equal volumes of water and 1M NH4C2H3O2. The precipitate will be analysed in Procedure 7 to isolate mercury from lead, bismuth, copper and cadmium.
Separation of mercury from lead, bismuth, copper and cadmium
We use warm dilute HNO3 to separate HgS, insoluble in its presence, from the sulphides of copper, cadmium and lead that are soluble: the dissolve in accordance with the equations
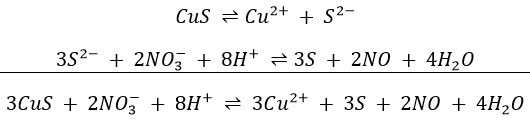
The second equation removes S2- from solution, shifting the first equation to the right.
Procedure 7
Add 15 drops of 3M HNO3 to the test tube containing the precipitate from Procedure 6, mix thoroughly, transfer to a casserole and boil gently for about one minute. Replenish the HNO3 when necessary. Transfer to a test tube, centrifuge, and decant in a test tube (save it for Procedure 8). Wash the precipitate (HgS and S) twice with 15-drop portions of water made acidic with one drop of 3M HNO3. Add the first washing to the decantate in the test tube but discard the second washing.
Treat the precipitate with 6 drops of 12M HCl and 2 drops of 16M HNO3, mix thoroughly and heat for 1 minute in the boiling water bath. Add 10 drops of hot water, transfer to a casserole, boil gently for 30s and then transfer back to a test tube. Cool by holding the test tube under the water tap, then centrifuge and decant into another test tube. Add 2-5 drop of 0.2M SnCl2 solution to the cool decantate in the test tube. A black (Hg) or grey (Hg2Cl2 + Hg) precipitates proves the presence of mercury(II).
Notes
1. If the HNO3 is too concentrated, it will dissolve some of the HgS. In addition, it may oxidise PbS to PbSO4, which will remain as a residue mixed with the HgS.
2. HgS is black. The presence of a white or yellow residue after digestion with HNO3 does not, however, eliminate the possibility of mercury being present. The residue, whatever its colour, should be tested for mercury.
3. HgS is insoluble in both concentrated HNO3 and concentrated HCl, when each is used separately. However, a mixture of the two concentrated acids dissolves HgS readily and quickly.
![]()
HgCl42- is pale yellow and is largely dissociated into colourless Hg2+ and Cl– ions when the solution is diluted with water. The Hg2+ ions can then be identified by means of the reaction written in note 4.
4. The extent to which Hg2+ is reduced by Sn2+ depends on the relative amounts of the two kinds of ions present. If Hg2+ is present in large excess, the reduction is mostly to Hg2Cl2:
![]()
Hg2Cl2 is white. If more Sn2+ ions are added, Hg2Cl2 that is first formed according to the reaction above is further reduced to Hg, as follows:
![]()
Hg is black. If Sn2+ is in excess, the following reaction takes place:
![]()
The combination of Hg2Cl2 (white) and Hg (black) forms a dark grey to black precipitate. The equations above show that tin(II) compounds are strong reducing agents.
The solution is boiled to remove chlorine formed by oxidation of Cl– by NO3–. The chlorine would oxidise Sn2+ to Sn4+, destroying the reductant (Sn2+) and preventing it from reducing the Hg2+.
Separation of lead from bismuth, copper and cadmium
Lead sulphate is insoluble in water. The sulphates of bismuth, copper, and cadmium are soluble. This fact is the basis for the separation of lead ions from the other cited ions.
Procedure 8
Add 4 drops of 18M H2SO4 to a casserole containing the decantate from Procedure 5 and evaporate under a hood until the volume is about 1 drop and dense white fumes of SO3 are formed. These fumes are dense and will obscure the view of the casserole inside walls. Paler fumes (from HNO3) must not be mistaken for the SO3 fumes.
Cool, add 15 drops of cold water, and stir the contents until all material in the casserole is dissolved or suspended; then transfer it to a test tube before the suspended material has a chance to settle. Swirl the casserole with 4 drops of cold water and transfer the washing to the same tube. Cool under the water tap. A white precipitate in the form of a fine suspension shows the presence of lead (PbSO4). Centrifuge until the supernatant liquid is clear and decant into a test tube (save for Procedure 9). Wash the precipitate twice with 10-drop portions of cold water. To the washed precipitate in the test tube, add 4 drops of 1M NH4C2H3O2 and stir for 20s. Then add 2 drops of 0.2M K2CrO4. A yellow precipitate confirms the presence of lead (PbCrO4).
Notes
1. PbSO4 is appreciably soluble in concentrated HNO3 due to formation of the hydrogen sulphate ion in the manner discussed in note 2. For this reason, HNO3 must be removed before PbSO4 will precipitate. When a solution containing H2SO4, HNO3 and water is boiled, the water and HNO3 are first driven off because they boil at comparatively low temperatures (100-120°C). Further heating results in boiling the H2SO4 (boiling point: 338°C). At its boiling temperature the H2SO4 decomposes to a slight extent.
![]()
SO3 fumes strongly in moist air. Therefore formation of dense white fumes of SO3 at the end of the evaporation gives assurance that all HNO3 has been removed.
2. When the solution is cooled down, sulphates of bismuth, copper and cadmium may crystallize out. However they are soluble in dilute H2SO4 and will dissolve if water is added. PbSO4, on the other hand is soluble in concentrated H2SO4 due to formation of HSO4–.
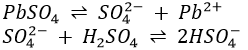
On dilution with water, the equilibria are shifted to the left and PbSO4 precipitates, precipitate finely divided while sulphates of bismuth, copper and cadmium form relatively large crystals.
3. PbSO4 dissolves the ammonium acetate because the complex ion Pb(C2H3O2)42-.
![]()
This complex ion is very stable and the solution contains thus a very low concentration of lead ions.
PbCrO4 is less soluble than PbSO4. Therefore, when K2CrO4 is added to the solution formed by adding NH4C2H3O2 to PbSO4, a precipitate of PbCrO4 is formed even though the concentration of lead ions in the solution is very low.
As lead is largely removed in the silver group, only very small quantities will ordinarily appear in the copper-arsenic group. For this reason, the test for lead may not be as strong as the test for other cations in this group.
Separation of bismuth from copper and cadmium
Addition of NH4OH to a solution containing bismuth, copper and cadmium ions first precipitates the hydroxides of all three metals. The hydroxides of copper and cadmium, however, dissolve in an excess of NH4OH. The hydroxide of bismuth does not.
Procedure 9
To the decantate from Procedure 8, add 15M NH3, dropwise and with constant mixing, until it becomes distinctly alkaline. Stir for 2 minutes. Centrifuge and decant (save it for Procedure 10). Wash the precipitate twice with 15 drops of hot water. To the washed precipitate, add 3 drops of 8M NaOH and 2 drops of 0.2M SnCl2 and stir. A jet-black precipitate proves the presence of bismuth (Bi).
Notes
1. When NH4OH is added to a solution containing copper ions, Cu(OH)2 (blue) is first precipitated.
![]()
An excess of NH4OH, however, dissolves the Cu(OH)2, to give a deep blue solution in which copper is present as the complex ion Cu(NH4)42+.
![]()
The detailed reaction is
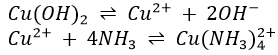
Cadmium behaves in the same manner as copper; Cd(NH3)42+ is formed. Cd(OH)2 (white) dissolves quite slowly in excess NH4OH; therefore the solution should be stirred for 2 minutes to ensure complete dissolution of Cd(OH)2. Similar complexes will be met in the case of the ions of nickel, cobalt and zinc. Ag(NH3)2+ was formed in the analysis of the silver group.
2. The formation of the black precipitate of bismuth results from the action of stannate(II) ions (Sn(OH)42-) on Bi(OH)3 (white):
![]()
The stannate(II) ions were formed when the SnCl2, added as the final reagent, reacted with the excess of NaOH.
![]()
The reaction of stannate(II) ions with Bi(OH)3 to give stannate (IV) and bismuth serves to illustrate the reducing character of tin(II) compounds. Stannate ions Sn(OH)42- are formed from the action of NaOH on SnCl2 in two steps. In a first time, if NaOH is added to a solution of SnCl2, a white precipitate forms:
![]()
If NaOH is put in excess, the white precipitate redissolves to form a clear, colourless solution containing the stannate ion.
![]()
If the white precipitate of Sn(OH)2 that first forms when NaOH is added is treated with an acid (such as HCl), it dissolves to form a clear solution that can be shown to contain Sn2+ ions.
![]()
Sn(OH)2 is thus amphoteric as is will dissolve in either a strong acid or a strong base. Amongst the metals we consider in our analyses, the hydroxides Sn(OH)2, Sn(OH)4, Pb(OH)2, Sb(OH)3, Al(OH)3, Cr(OH)3 and Zn(OH)2 are amphoteric, and the other are not amphoteric.
It should be pointed out that there is not complete agreement amongst chemists about the composition of the stannate ion: Sn(OH)42-, SnO22-, Sn(OH)3– and HSnO2– are suggested. Since the formula of the ion may change as the concentration of the solution changes, it may well be that all four are present at one time or another, or even may all be present in equilibrium at one time.
Stannate ions reduce the hydroxides of antimony, lead, copper and cadmium to the corresponding metal, but the process is slow and the metallic deposit is not jet-black. Bi(OH)3 is also reduced but it forms instantly a jet-black deposit of metallic bismuth.
Finally, the stannate(II) ions react in contact with air and with itself to form stannate(IV) ions.
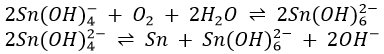
For these reasons, stannate(II) is not kept as a reagent but is formed at the time it is to be used.
Detection of copper and cadmium
Procedure 10
Detection of copper:
If the decantate from Procedure 9 is colourless, copper may or may not be absent: solutions containing copper in amounts less than 1 part in 25000 may appear colourless. If the decantate is deep blue, copper is present (Cu(NH3)42+. It is wise to verify the presence of copper in either case. To do so, place 5 drops of the decantate (not all the decantate, cadmium may be present) in a test tube and add 5M HC2H3O2 until the solution is decolorized. Then add 2 drops of 0.2M Fe(CN)6. A red precipitate confirms the presence of copper (Cu2Fe(CN6).
Detection of cadmium:
If copper is absent, treat the colourless decantate from Procedure 9 with 2 or 3 drops of ammonium sulphide solution, mix thoroughly, and allow to stand for about one minute. The formation of a yellow precipitate proves the presence of cadmium (CdS).
If copper was confirmed before, add 0.2M KCN (caution: KCN is poisonous) dropwise to a 10-drops portion of the blue decantate until the colour disappears. Then treat the solution with 2 or 3 drops of ammonium sulphide solution, mix thoroughly and allow to stand for about one minute. A yellow precipitate proves the presence of cadmium (CdS). As soon as the test is completed, dump the contents of the tube to which KCN was added into the sink and flush it away with a lot of water.
Notes
1. The blue colour of Cu(NH3)42+ is visible as little as 1 part of copper is present in 25000 parts of water. The red precipitate of Cu2Fe(CN)6 will detect 1 part of copper in 1 million parts of water.
2. Cu2Fe(CN)6 and CdFe(CN)6 are soluble in strong acids (HCl and H2SO4) but precipitates readily in presence of a weak acid (such as acetic acid).
3. KCN is poisonous and is thus not kept available as usual reagents, because of potential accidents. It must never be mixed with an acid or an acid solution. Even a very weak acid will react with it and liberate poisonous HCN gas
4. When excess KCN is added to a solution containing Cu(NH3)42+ and/or Cd(NH3)42+, the complex ions Cu(CN)2– and/or Cd(CN)4– are formed. Cu(CN)2– is very stable and only very slightly dissociated into Cu+ and CN–. The concentration of Cu+ is so low that we don’t obtain any precipitate of Cu2S when sulphide ions are added. Cd(CN)42- is less stable and dissociates appreciably into Cd2+ and CN–. The concentration is high enough for a precipitate of CdS to form when sulphide ions are added.
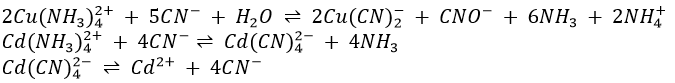
Reprecipitation of the sulphides of arsenic, antimony and tin
Procedure 11
To the test tube that contains the decantate from Procedure 6 (potentially containing AsS43-, SbS33- and SnS32-), add 6M HCl under a hood with constant stirring until the solution shows an acidic reaction when tested with litmus. As long as each drop of 6M HCl keeps on bringing down more precipitate, the solution is still alkaline; when no more precipitate forms, the solution is probably acidic. A large excess of HCl must be avoided (see notes). Centrifuge and decant, discarding the decantate. Wash the precipitate three times, each time with 10 drops of a hot solution prepared by mixing equal volumes of water and 1M NH4C2H3O2, and analyse according to Procedure 12.
Notes
1. A dark-coloured decantate from Procedure 6 means that some CuS, and perhaps HgS, has been put into a state of colloidal suspension by the (NH4)2 If this decantate is allowed to stand for about 24h, the CuS and HgS will ordinarily settle out. The yellow supernatant liquid can then be decanted, leaving the CuS and HgS behind. If the decantate is to be analysed immediately, add 5 drops of 1M NH4C2H3O2 to coagulate the CuS and HgS, mix thoroughly, centrifuge, and decant, discarding the precipitate.
2. The addition of dilute HCl to the soluble thiol salts of the arsenic subgroup reprecipitates the sulphides of the three elements in accordance with the following equation
![]()
The compound H3AsS4 is unstable and decomposes.
![]()
(NH4)2SnS3 and (NH4)3SbS3 react in a similar manner. H2SnS3 and H3SbS3 decompose to give SnS2 and Sb2S3, respectively.
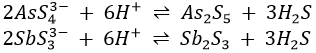
H2S plays thus the same role as H2O and As2S5, Sb2S3 and SnS2 are the equivalents of acid anhydrides in the sulphur acid system.
3. In reprecipitating the arsenic group, a large excess of HCl must be avoided, since SnS2 and Sb2S3 are appreciably soluble in high concentrations of this acid. Also, the liquid in contact of their precipitates should be decanted off at once since the SnS2 and SbS3 will dissolve on long standing.
4. The precipitate that is formed in Procedure 11 may be only sulphur. Ammonium sulphide may contain some ammonium polysulfide (NH4)2S2. When HCl is added to a solution containing it, sulphur is liberated according to the reaction
![]()
Since (NH4)2S2 is a moderately strong oxidizing agent, it will oxidize As(III) to As(V) in Procedure 6.
![]()
Separation of arsenic from antimony and tin
Arsenic sulphide is insoluble in concentrated HCl. As written in the note 3 above, sulphides of tin and antimony dissolve in the HCl to form soluble chlorides.
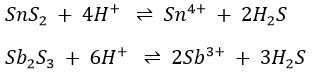
The Sn4+ and Sb3+ ions react with the Cl– ions present to form the complex chlorous ions SnCl62- and SbCl4–.
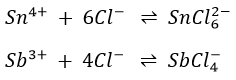
Procedure 12
To the precipitate from Procedure 11, add 15 drops of 12M HCl, mix thoroughly and heat the test tube in the boiling water bath for 3-4 minutes, stirring frequently. Add 7 drops of hot water, mix well and continue the heating for 20-30s. Centrifuge and decant into a test tube. Save the decantate (for Procedure 14) and wash the precipitate once with 10 drops of 6M HCl and then 3 times with 10-drops portions of a hot solution prepared by mixing equal volumes of water and 1M NH4C2H3O2. Analyse the precipitate according to Procedure 13.
Note
- The precipitate (As2S5) is first washed with HCl rather than with water to remove all traces of Sb3+ because they will react with water to form insoluble SbOCl, which will remain with the As2S5. The final washing with hot water is to remove all traces of HCl. Chloride would interfere with the confirmatory test for arsenic (Procedure 13) by forming AgCl.
Detection of Arsenic
Procedure 13
Add 10 drops of 16M HNO3 to the test tube containing the precipitate from Procedure 12 and heat in the boiling water bath for one minute, or until the original precipitate is disintegrated and a deposit of sulphur is formed. Add 4-5 drops of water, centrifuge, and decant into a casserole, discarding any precipitate that remains in the test tube. Evaporate the solution in the casserole very carefully to complete dryness, but do not bake. Allow to cool. Add 4 drops of 0.2M AgNO3 and swish around in the casserole for 10 seconds. A reddish brown precipitate proves the presence of arsenic (Ag3AsO4). If no reddish brown precipitate appears, add 0.2M NaC2H3O2, a drop at a time and with thorough mixing, until precipitation is complete or a maximum of 30 drops has been added. A reddish brown or chocolate brown precipitate proves the presence of arsenic.
Notes
1. As2S5 and As2S3 are dissolved by HNO3 this way:
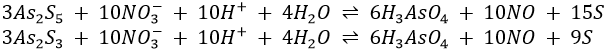
If the H3AsO4 is heated to 160-200°C, As2S5 is formed.
![]()
Ag3AsO4 is reformed when the aqueous solutions of AgNO3, and NaC2H3O2 are added.
![]()
2. Ag3AsO4 is formed in the confirmatory test for arsenic according to the equation
![]()
Ag3AsO4 is the salt of a weak acid and is, therefore, soluble in HNO3. However, it is insoluble in a weakly acidic or neutral solution. Evaporation to dryness should remove all trace of HNO3. As a precaution against the possibility of some HNO3 being left, NaC2H3O2 is added to the reaction mixture. As the salt of a weak acid, NaC2H3O2 will serve as a buffer for any HNO3 that might have remained. The C2H3O2– ions from the strong electrolyte NaC2H3O2 combine with H+ ions from HNO3 to form the weak acid HC2H3O2. This reaction maintains the H+ ion concentration at such a low value that Ag3AsO4 will precipitate.
3. The formation of a white precipitate (AgCl) means that the As2S5 was not washed free of chloride in Procedure 12. AgC2H3O2 (white) is sparely soluble but may also precipitate.
Detection of antimony and tin
Procedure 14
A. Detection of antimony
Transfer the decantate from Procedure 12 to a casserole, boil for 1 min to remove all H2S, then add 4-5 drops of cold water and mix thoroughly. Place 4 drops of this solution in a test tube and add 5M NH4OH drop by drop with constant stirring until the solution is barely alkaline (showed by a litmus test).
If antimony or tin is present, a white precipitate or milky suspension, SbOOH and/or Sn(OH)4 will form in the alkaline solution. If there is no precipitate (or suspension), then tin and antimony are absent (no need to continue Procedure 14 then).
Add 5MHC2H3O2 drop by drop with constant stirring until the solution is acidic to litmus. The white precipitate should remain in the weakly acidic solution and does not interfere with the test for antimony. Add one crystal of Na2S2O3•5H2O and heat by placing the test tube in the boiling water bath for 2-3 min. An orange red precipitate proves the presence of antimony (Sb2OS2).
B. Detection of tin
To the remainder of the solution in the casserole, add a 1-in. piece of 26-gauge aluminium wire. Warm gently until the wire has dissolved; then boil gently for about 2 minutes, or until the black precipitate either has all dissolved or appears not to be dissolving anymore, replenishing the solution with 6M HCl if necessary. If the black precipitate dissolved completely, then antimony is absent. If after 2 minutes there is still a black residue, Sb is present. Transfer the contents of the casserole immediately to a test tube, cool, centrifuge and decant.
Immediately add 2-3 drops of 0.1M HgCl2 solution to the decantate, mix thoroughly and allow to stand one minute. The presence of tin is showed by the presence of a white (Hg2Cl2) or grey (Hg2Cl2 + Hg) precipitate.
Notes
1. The solution used in making the confirmatory test for antimony must be free from H2S; otherwise SnS2, as well as Sb2S3, will precipitate when the middle is made alkaline.
2. Addition of NH4OH precipitates SbOOH and Sn(OH)4 according to the following equations:

The detailed reaction are between the Sb3+ and Sn4+ ions in equilibrium with SbCl4– and SnCl62- and the OH– ions in equilibrium with NH4OH.
3. The thiosulfate ion S2O32- disproportionates sparingly to yield H2S.
![]()
Although the concentration of S2- ions provided by this H2S is not high enough to precipitate tin as SnS2, it will precipitate antimony as Sb2OS2.
![]()
4. A flame test for tin may be carried out as follows: use cold water to fill a clean (outside and inside) test tube. Dip the bottom of the tube into the solution to be tested; then hold the bottom of the tube in a hot, non-luminous Bunsen flame. A blue coloration of the flame, which appears to cling to the wall of the test tube, proves the presence of tin. A trial flame test for tin should first be run on a sample of tin salt solution from the reagent shelf so that the characteristic blue coloration can be recognized.
5. The aluminium is added to reduce Sn4+ to Sn.
![]()
Excess aluminium is dissolved by the HCl. The tin then dissolves in HCl to form Sn2+ ions.
![]()
The tin does not dissolve until all the aluminium has dissolved or the excess is removed. Furthermore, since tin is not very active, it dissolves less readily than the aluminium. For that reason the mixture must be boiled.
Since the tin(II) formed by the reaction of tin with HCl may eventually be oxidised by the oxygen of the air to tin(IV), the test should be completed as rapidly as possible.
6. The chemistry of the final test for tin, in which Sn2+ reduces Hg2+ to Hg+Hg2Cl2, is the same as that involved in the confirmatory test for mercury (Procedure 7, note 4).
The metallic aluminium added to the solution containing Sn4+ and Sb3+ replaces antimony (black), as well as tin (grey). Antimony, being less active than hydrogen, does not dissolve in HCl however.