The primary structure of a protein is the succession, or sequence, of the AA. Proteins are made of one linear chain of amino acids. This linear chain takes a 3D structure (secondary and tertiary structures) because of the interactions between the sequences in the chain and the interactions (hydrophobic or hydrophilic) with the surrounding. The order of the AA and their quantities are well defined for each protein. The first structure ever determined was the one of the insulin. This protein is 51 AA long, what is small in comparison with other proteins that can be up to 8000 AA long.
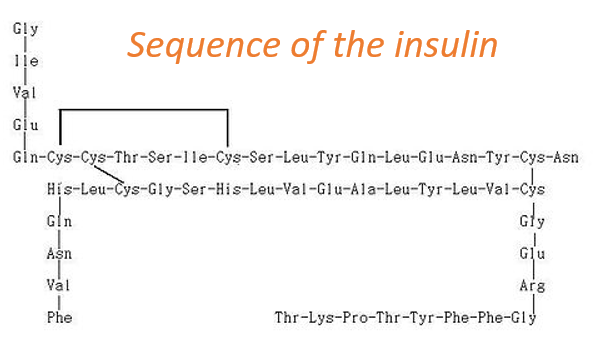
At the exception of the disulphide bridges between the cysteines, this protein may be seen as two linear chains with respectively 21 and 30 AA. As the insulin is not composed of one single chain, this protein is obviously not the primary product of a ribosome. Some further operations were applied to the precursor forms of the insulin to obtain the functional protein. We will see this process in more details later.
To determine the primary structure of a protein, we need it to be pure. To separate the proteins from the other molecules, we perform a dialyse: the solution containing the proteins in a bag with a porous membrane through which small ions can pass but not macromolecules. Once the equilibrium is reached, we repeat the process to obtain the best purity possible. We still have to separate macromolecules. It is done
- by chromatography
- ionic: the proteins have a charge that depends on the R of the amino acids. The charge depends on the solvent that is used. At one given pH, the target protein is positively charged. Placing a polymer negatively charged in the column will retain the protein while the others pass through the column. After a while we change of solvent to unbind the protein from the polymer.
- of exclusion: the proteins are separated as a function of their size. The large protein move faster than the small ones because they cannot enter in the holes of the beads composing the immobile phase.
- ligands: the protein can bind with specific ligands. Normally only this protein should bind, what assures a great purity.
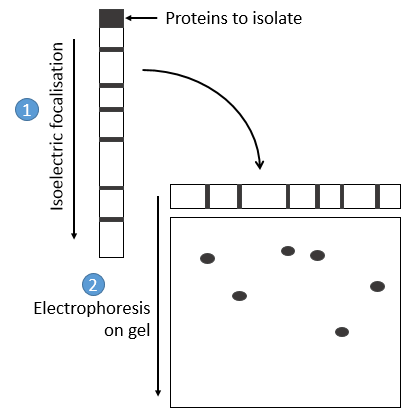
- by electrophoresis: the proteins are separated on the basis of their isoelectric point: the proteins are placed on a gel of polyaccrylamine the pH of which is not uniform but a gradient. As the proteins are charged in function of the pH of the solvent, it will move until its charge becomes neutral. After that, we take the gel and apply it on top of a second, long porous gel where the proteins are displaced by the application of a charge and their speed also depends on the size of the protein. This way we obtain a 2D separation of the species.
Once the proteins are isolated, we can start to determine its structure. To do so, we use an exopeptidase: its role is to cleave only the liaison before the last AA. The problem is that the enzymes are not synchronic: they don’t all start simultaneously and some chains may be cleaved several times while others are cleaved once or not at all. There can thus be a mix of amino acids in the solution and the determination of the right sequence may be difficult. To minimize this problem, the experiment has to be very short.
Another method (of Edman) is to weaken the last peptide bond and next to use a weak reactant that hydrolyses only this liaison. In the presented case, a phenyl isothiocyanate reacts with the amino-terminal residue to form a thioamide. The next peptide bond is weakened and can be cleaved without hydrolysis because of the formation of a cycle.
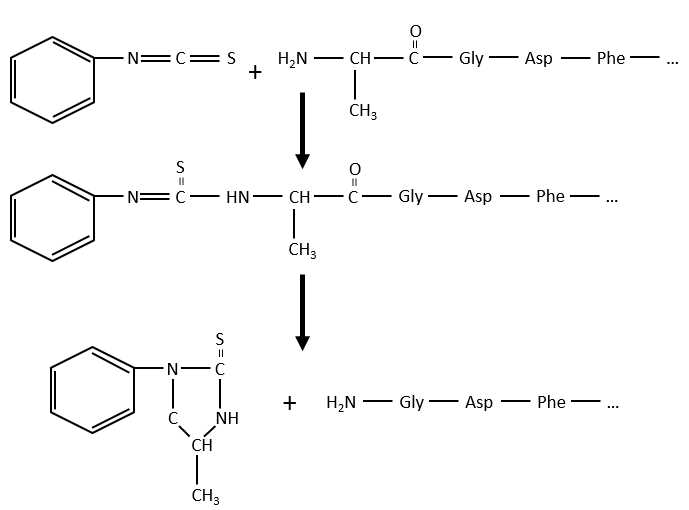
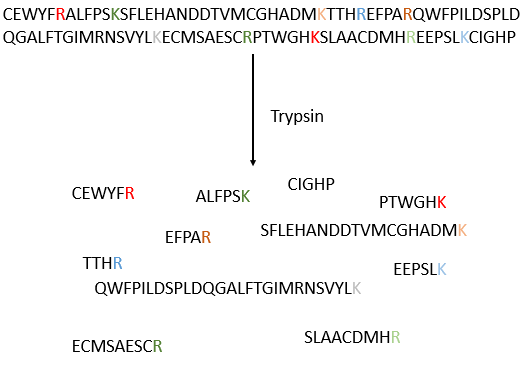
The process can be repeated several times to determine the initial amino-terminal sequence but the yield of the reaction is not of 100%. As a result, it can only be repeated around 30 times. Moreover, sometimes chains break and we have thus a new extremity that gives different results. Anyway 30 AA is not good enough to determine the structure of proteins that can be 8000 AA long. The order of the 30 first amino acids don’t give any clue on the nature of the following amino acids. The only way to determine the structure is to first fragmentise the protein into small bits. We will try to cleave the protein only between some amino acids to have points of reference. There are some specific endopeptidases enzymes that cleave proteins only after one particular amino acid. It detect the presence of one specific R, binds to it and cleaves the peptide bond. The trypsin is one enzyme that cleaves the peptide bond after one lysine or arginine.
We separate the fragments by chromatography and determine the sequence of each fragment (with the method of Edman for instance). Yet, we don’t know in which order the fragments are in the protein. The process is repeated with another specific endopeptidase. After several fragmentations and sequencings, we can try to figure out which sequences are in common in different fragmentations and reconstruct the whole sequence.
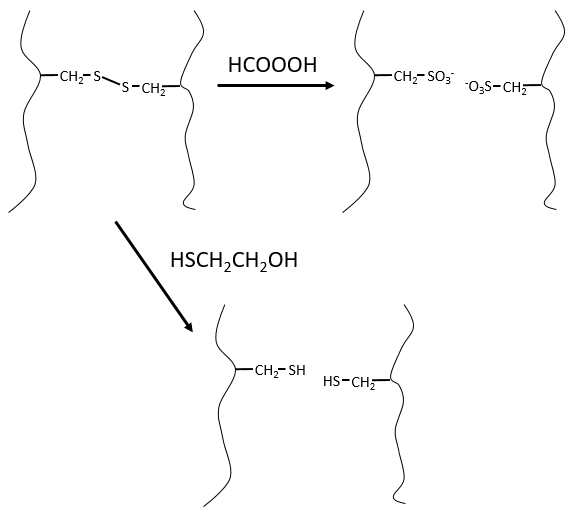
It would be perfect if there were no SS bonds along the peptide chain. There is thus a step that has to be done prior to the fragmentation: we use performic acid (HCOOOH) to oxidise the sulphurs. This method is irreversible. A second method, reversible, can be used instead: the mercaptoethanol reduces the sulphurs.
We can resume the primary structure in 4 points:
- the chain is linear and non-branched.
- between the amino acids, the only covalent bonds are the peptide bonds and the SS bonds between the cysteines. The other interactions are attraction/repulsion between charged groups and H bonds.
- the sequence is completely defined and arbitrary: one part of the sequence does not defines the rest of the sequence. For instance, if we know 10 amino acids, we cannot say that the 5 next are a guanine, a cysteine, etc…
- proteins from different species are similar but not identical. There are some key fragments that are common but the whole structure may vary. One exception is the histone. This protein is common to all the eukaryote species: it is the protein responsible for the compaction of the DNA in the cellular nucleus. Even if the information contained in the DNA differs between species, its structure is identical and the protein responsible for its coiling/uncoiling is thus the same for all the eukaryote species.
Some proteins fix one (or more) monosaccharide. This process is called glycosylation and lead to one glycoprotein.
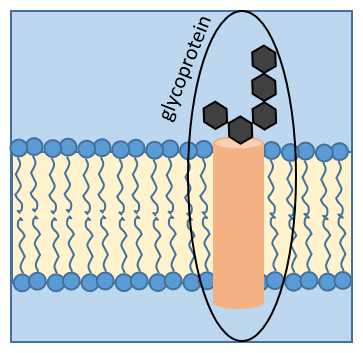
The monosaccharide can be fixed on one nitrogen atom (N-linked glycosylation, exclusively on the asparagine) or one oxygen atom (O-linked glycosylation). The monosaccharide can itself be bound to a chain of monosaccharides. We frequently observe glycoproteins in membranes where they play the role of receptors. The presence of the monosaccharide on the protein permits a great diversity in the receptors for the cells.